
Krátký úvod do ChIP-Seq

Interakce protein-DNA jsou široce využívány k objasnění mechanismů, které jsou základem buněčné fyziologie. Vývoj technologie chromatinové imunoprecipitace (ChIP) umožnil studium těchto mechanismů. Po dalším vývoji vznikla alternativa hloubkového sekvenování (ChiP-Seq), která nabízí výhody z hlediska specifičnosti a citlivosti.
Experiment ChIP-Seq začíná zesíťováním celé buňky formaldehydem, následuje sonikace a izolace DNA. Poté se provede imunoprecipitace komplexu DNA-protein, který se skládá z protilátek vážících se na specifické proteiny. Vzniklé imunokomplexy se vysráží a přečistí. Nakonec se DNA sekvenuje, čímž se získají údaje o obohacených místech s vysokým rozlišením. Tento přístup spolu s dobře zavedenou pipeline ChIP-seq umožňuje výzkumníkům zachytit DNA transkripční faktory, místa modifikace histonů, epigenetické změny a podpisy genových regulačních sítí.
Klinický význam a aplikace
Epigenetické nerovnováhy napříč nemocemi a zdravotními stavy mohou zahrnovat modifikaci histonů a změněné transkripční faktory. Zde byly studie ChIP-Seq použity k objasnění patologických molekulárních mechanismů, které jsou základem rakoviny a dalších onemocnění. Analýza ChIP-seq také přispívá k pochopení úlohy transkripčních faktorů během onemocnění. Zdá se totiž, že některé transkripty jsou během klinických fenotypových projevů změněny.
Přehled pipeline ChIP-Seq
Pipeline analýzy ChIP-Seq je hlavní součástí projektů interakce DNA s proteiny a skládá se z několika kroků, včetně zpracování surových dat, analýzy kontroly kvality, zarovnání k referenčnímu genomu, kontroly kvality zarovnaných čtení, volání píků, anotace a vizualizace. Pro získání vysoce kvalitních výsledků v experimentu ChIP-seq je však zásadní mít promyšlený experimentální design. Před zahájením analýzy je nezbytné zvážit parametry, jako jsou replikáty vzorků, kontrolní skupiny, sekvenační soupravy a sekvenační platformy.
Kontrola kvality
Všechny zprávy Basepair poskytují skóre kvality, které pomáhá odhalit potenciální problémy se sekvenováním nebo kontaminaci vstupních dat.
Krok kontroly kvality (QC) má za cíl vyhodnotit kvalitu vysoce výkonných dat získaných sekvenováním. Tento krok je podobný těm, které se provádějí při analýzách DNA-seq a RNA-seq. Zde mezi hlavní hodnocené metriky patří kvalita sekvencí a bází, obsah GC, přítomnost sekvenačních adaptérů a nadměrně zastoupených sekvencí. Jedním z nejčastěji používaných programů pro tento typ analýzy je FastQC. Pokud jsou navíc identifikovány sekvence s nízkou kvalitou, lze je později odstranit během kroku ořezávání. Ačkoli se jedná o nepovinný krok, trimování zlepšuje kvalitu dat tím, že zachovává pouze vysoce kvalitní čtení.
Zarovnání
Po měření QC jsou čtení ChIP-Seq zarovnána k referenčnímu genomu. Mapování čtení umožňuje výzkumníkům určit původ sekvence čtení v genomu. Mezi oblíbené používané softwarové nástroje pro zarovnávání patří Bowtie a BWA, které se používají v pipelinech ChIP-seq společnosti Basepair. Oba nástroje mapují málo odlišné sekvence oproti referenčnímu genomu.
Průtok počtu čtení pomáhá poskytnout celkový přehled o použitelných čteních na konci procesů ořezávání, zarovnávání a deduplikace. Představte si obrázek jako montážní linku analýzy dat: vstupní surová data, výstupní použitelná čtení.
Kontrola kvality zarovnaných čtení
Další krok spočívá v odvození kontroly kvality zarovnané sady dat. Během procesu mapování způsobují duplicity čtení zavedené amplifikací PCR a sekvenováním zkreslení při volání píků a analýze obohacení. Basepair používá k odstranění duplicit nástroj Picard. Po odstranění duplikátů byste měli vyhodnotit neredundantní frakci (NRF) zarovnaných čtení. NRF měří unikátní čtení mapující referenční genom. Ideální experimenty ChIP-seq by měly mít méně než tři čtení na pozici.
Krok vyvolání vrcholu
Krok vyvolání vrcholu detekuje obohacené oblasti interakce protein-DNA v genomu. Pipeline Basepair ChIP-seq používá k provedení této analýzy MACS2. V MACS2 se volání píků provádí na základě tří hlavních kroků: odhad fragmentů, následuje identifikace lokálních parametrů šumu a poté identifikace píků. Jako výstup tohoto kroku uživatelé získají konečnou tabulku s informacemi o píku, jako je skóre obohacení, hodnota -log10p, hodnota -log10q a pozice k začátku píku. V tomto kroku se důrazně doporučuje použít kontrolní vzorky pro porovnání se zkoumaným cílovým souborem dat. Mějte na paměti, že dobré kontrolní skupiny přinášejí spolehlivější výsledky.
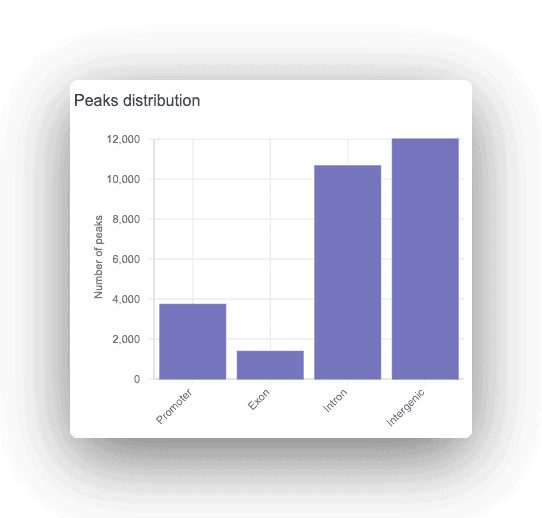
Každý pík je anotován jako promotorový, intronický nebo intergenní se zobrazením příslušného genu. U všech nalezených píků se provede analýza motivů, aby se našla nadreprezentovaná vazebná místa transkripčních faktorů.
Přehled výsledků
Pipeline ChIP-seq může poskytnout nejen informace o stavu chromatinu, ale také o vazbě transkripčních faktorů v určeném kontextu genu nebo lokusu. Výskyt modifikací histonů a transkripčních faktorů v regulačních oblastech DNA může představovat epigenetický podpis specifický pro daný stav. Epigenetické poruchy tak mohou být spojeny s klinickými fenotypy. Například heterogenita stavů chromatinu může vést k rezistenci na léčbu u karcinomu prsu. Tyto buňky mají tendenci ztrácet markery represivních modifikací histonů a dále zvyšovat expresi genů, o nichž je známo, že podporují rezistenci k léčbě rakoviny.
Analýza vrcholů, motivů a cest v pipeline analýzy ChIP-Seq
Identifikace obohacení transkripčních faktorů o motivy se používá k objasnění toho, zda transkripční faktory v dané oblasti spolupracují, nebo si konkurují. Identifikace vrcholů v oblastech s motivy DNA může zlepšit interpretaci experimentálních výsledků. Analýzy vrcholů i motivů společně umožňují nahlédnout do toho, co se může v buňce odehrávat. Výsledkem integrace obohacení píků a motivů je epigenomická krajina s možnými biologickými důsledky. Dále se analýza drah používá k identifikaci proteinů v dráze. Vyšetřování a závěry jsou formulovány na základě přítomnosti proteinů.
Vizualizace dat
Výsledná data z pipeline ChIP-seq lze vizualizovat pomocí prohlížeče genomu. Zprávy Basepair obsahují vestavěný prohlížeč genomu IGV2, který umožňuje interakci s vámi získanými daty. Data lze alternativně vizualizovat pomocí heatmaps, což jsou infografiky reprezentativní intenzity založené na hustotě dat, které ukazují přítomnost nebo nepřítomnost konkrétních značek. Mezi další zde používané grafiky patří graf obohacení, upSet a graf pokrytí, který vypočítává a zároveň zobrazuje pokrytí špičkových oblastí v celém genomu.
Genomový prohlížeč je skvělým nástrojem pro vizualizaci vašich surových genomových dat. Je zabudován do každé zprávy o analýze ChIP-seq v aplikaci Basepair.
1. Grosselin, K., A. Durand, et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet, v.51, n.6, Jun, p.1060-1066. 2019.
2. Northrup, D. L. e K. Zhao. Použití ChIP-Seq a příbuzných technik ke studiu imunitních funkcí. Immunity, v.34, č.6, 24. června, s.830-42. 2011.
3. Park, S. J., J. H. Kim, et al. A ChIP-Seq Data Analysis Pipeline Based on Bioconductor Packages. Genomics Inform, v.15, č.1, březen, s.11-18. 2017.
4. Pepke, S., B. Wold, et al. Computation for ChIP-seq and RNA-seq studies. Nat Methods, v.6, n.11 Suppl, Nov, s.S22-32. 2009.
5. Satoh, J., N. Kawana, et al. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul Syst Bio, v.7, p.139-52. 2013.