
Úvod
Hypokalémie je častým klinickým problémem v praxi endokrinologů a nefrologů. Existuje mnoho zjevných příčin hypokalémie, jako je průjem, zvracení nebo zneužívání diuretik. Jiné příčiny, jako jsou tubulopatie, se vyskytují zřídka a jejich diagnostika je náročnější. Existuje mnoho dědičných i získaných tubulopatií způsobujících hypokalémii, někdy závažnou a život ohrožující.1
Relativně častou, ale přehlíženou příčinou hypokalémie je Gitelmanův syndrom (GS).2 Jedná se o recesivní tubulopatii ztrácející soli způsobenou mutací genu SLC12A3. Gen SLC12A3 kóduje thiazid-senzitivní transportér NCCT (sodium chloride co-transporter). NCCT se nachází v distálních stočených tubulárních buňkách (DCC), které jsou zodpovědné za 7-10 % tubulární absorpce elektrolytů.3
Nejzávažnějšími laboratorními abnormalitami zjištěnými u GS jsou hypokalémie a hypomagnezémie způsobené renálním úbytkem K+ a Mg2+. Dalšími typickými změnami jsou metabolická alkalóza, hypokalciurie a hyperreninemický hyperaldosteronismus.4 Často je pozorována mírná až středně těžká hypofosfatemie.5 Byla zaznamenána i těžká hypofosfatemie s těžkou hyponatremií.6,7
První příznaky GS se objevují u dětí nebo mladých dospělých s normálním růstem a anamnézou chuti na sůl (děti dychtivě konzumují kyselé okurky nebo slaný nálev, solené okurky, pomeranče a citrony, děti olizují sůl z bramborových lupínků atd. „8 Klinický obraz se u jednotlivých pacientů liší. Někteří jsou asymptomatičtí, ale u jiných se vyvinou život ohrožující komplikace. U mužů se projevuje závažnější fenotyp než u žen.8 Nejčastějšími příznaky jsou svalové křeče a slabost, zácpa, nokturie, polyurie, žízeň, polydipsie, srdeční arytmie, parestézie a zvýšená chuť na sůl. Arteriální hypotenze je častá a v mnoha případech je nejvýraznějším příznakem, u stárnoucí populace GS se však může objevit hypertenze.8 Korelace mezi biochemickými abnormalitami a příznaky není silná.9 GS nenarušuje náladu a sociální vztahy dětí.9,10 Jinak jsou příznaky častější u dospělých a mohou mít negativní dopad na kvalitu jejich života. Čtyřicet pět procent pacientů s GS považuje své příznaky za střední až velký problém.11 Extrémní vyčerpání, svalová slabost, parestézie, silná únava a hypotenze jsou spojeny s mírným až silným omezením každodenních aktivit.9
Odhadovaná prevalence GS je 1:40 0008 a prevalence heterozygotů je v evropské populaci nejméně 1 %. Dosud bylo popsáno více než 180 různých mutací v SLC12A3.12
Popis případu
26letý muž byl přijat do nemocnice kvůli výskytu synkop, generalizované a svalové slabosti a svalových křečí. V anamnéze pacienta byla zjištěna epizoda synkopy s hladinou draslíku 3,16 mmol/l. Při dalším sledování v ambulantním hodnocení byl pozorován opakovaný výskyt hypokalémie (nejnižší hodnota 2,6 mmol/l). Krevní tlak byl v normě 110/80, srdeční frekvence 72 za minutu; při fyzikálním vyšetření nedošlo k žádným změnám. Ani neurologický nález, hmotnost 74 kg, výška 178 cm. Při přijetí do nemocnice laboratorní vyšetření prokázalo mírnou hypokalémii (K+ 3,0mmol/l), hypomagnezémii (Mg2+ 1,36mg/dl), hypokalciurii (
mg/24h) a metabolickou alkalózu (HCO3- 29,7mmol/l, BE 5,3mmol/l). Funkce ledvin byla dobrá s eGFR>60ml/min. Zobrazovací vyšetření byla bez pozoruhodností, stejně tak EKG.
Další vyšetření potvrdila, že hypokalémie byla způsobena renálním úbytkem draslíku. Hodnoty 24hodinového plýtvání draslíkem, transtubulárního gradientu K+ (TTKG), frakční exkrece K+ (FeK) a náhodného poměru K/kreatinin (K/Cr) byly typické pro hyperkaliurii a jsou uvedeny v tabulce 1.13-15 Denní magneziurie byla 64 mg/24 h, přičemž frakční exkrece hořčíku (FeMg) byla 15 %.
Hodnoty diagnostické pro hypokalémii.13-15
| Hodnoty diagnostické pro nepřiměřenou kaliurézu | Pacient | |
|---|---|---|
| Močový K+ | >20mmol/24h Jestliže sérový K+3.5mmol/l |
57mmol/l (sérový K+=3.0mmol/l) |
| FeK=×100% | >6,5% Pokud je sérový K+3.5mmol/l |
12% (sérový K+=3,0mmol/l) |
| TTKG= | TTKG>2 Jestliže sérový K+3.5mmol/l |
7,3 (sérový K+=3,0mmol/l) |
| uK/Cr – náhodný poměr K+ a kreatininu v moči | uK/Cr>15mmol/g Jestliže sérový K+3.5mmol/l |
67mmol/g (sérový K+=3.0mmol/l) |
Přestože byl krevní tlak normální, byly provedeny hormonální testy k vyloučení Connovy choroby a CT břicha, které neodhalily žádné abnormality v nadledvinách. Pro GS byl typický sekundární hyperaldosteronismus s hladinami aldosteronu 289pg/ml (normální rozmezí 20-180) a reninu 205mIU/ml (normální rozmezí 2,8-39,9).
Pacient byl diagnostikován jako GS na základě klinického fenotypu. Po stanovení diagnózy byla zavedena léčba doplňky draslíku a hořčíku. Dostával 20 mmol chloridu draselného a 18 mmol pyrolidonkarboxylátu hořečnatého, přičemž došlo ke klinickému zlepšení. V rodinné anamnéze byla zjištěna mírná asymptomatická hypokalémie u 35leté sestry pacienta. Vyšetření však neprokázala hyperkalurii ani hypomagnezémii. GS nebyl v tomto případě diagnostikován.
Genetická analýza
Podle genetické analýzy, algoritmu navrženého Nozu16SLC12A3 gen by měl být testován u pacientů s hypokalemickou metabolickou alkalózou, s plnohodnotným porodem, normální hmotností bez nefrokalcinózy, s hypokalcurií a hypomagnezémií.
V popsaném případě bylo provedeno genetické vyšetření a byly zjištěny dvě heterozygotní mutace: c.35_36insA a c.1095+5G>A v transkriptu NM_000339.2 genu SLC12A3 (obr. 1). Obě mutace zatím nejsou v dostupné verzi databáze HGMD(r) . První mutace byla nalezena také u matky pacienta a druhá u otce. Pouze jedna ze dvou mutací identifikovaných u našeho pacienta c.35_36insA byla nalezena u jeho sestry.
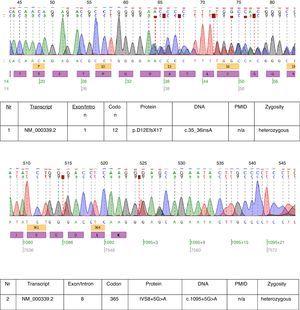
Chromatogramy sekvenování DNA postiženého pacienta.
Mutace nalezené u tohoto pacienta téměř dokazují klinickou diagnózu GS. Patogenetický význam mutace s posunem rámce je zřejmý. Patogenetickým významem mutace v místě sestřihu intronu 8 si nemůžeme být jisti. Konsenzuální sestřihová sekvence G (100 %), T (100 %), A (62 %), A (68 %), G (82 %), T (63 %) u dárce sestřihu intronu 8 neodpovídá přesně konsenzu GTgAGc. Mutace nalezená u tohoto pacienta dále odchyluje sekvenci od konsenzu (GtgAac). Také analýza in silico pomocí testeru mutací považuje tuto mutaci za způsobující onemocnění.17
Diskuse
Tubulopatie jsou vzácná onemocnění. Podle databáze RenalTube18 jsou nejčastějšími primárními tubulopatiemi v evropské populaci distální renální tubulární acidóza, Bartterův syndrom, familiární hypomagnezémie s hyperkalciurií a GS.18 Prevalence GS se pohybuje kolem 25 případů na 1 milion obyvatel.3 Protože GS patří mezi nejčastější, pravděpodobně se s případy GS během své kariéry setká mnoho nefrologů a endokrinologů. Náš pacient měl typický klinický obraz GS a dobře reagoval na léčbu. Problémem GS je, že přehlédnutá hypokalémie může způsobit smrt v důsledku srdeční zástavy nebo ochrnutí dýchacích svalů. Závažný nervosvalový příznak, jako je hypokalemická paralýza, se vyskytuje až u 6 % pacientů (častěji u asijských pacientů).3
Dědičné tubulopatie mají jakýsi „zrcadlový obraz“ – získané tubulopatie způsobené diuretiky. Každé diuretikum (přesněji: natriuretika, akuretika nebo glukuretika) způsobuje abnormality podobné těm, které se vyskytují u dědičných tubulopatií (tabulka 2). Téměř u každé tubulopatie se vyskytuje polyurie a změny hladiny draslíku patří mezi nejčastější problémy. U pacientů s GS se vyskytují mutace genu SLC12A3. Tento gen kóduje thiazid-senzitivní transportér. Proto se GS podobá chronické léčbě thiazidy.
Diuretika a tubulopatie.
| Dědičný defekt jednoho genu | Koncentrace draslíku | Příznaky podobné užívání: | Gen | Genový produkt | Umístění genového produktu |
|---|---|---|---|---|---|
| Guibaud-Vainselův syndrom (RTA 3) | ↓ | Acetazolamid | CA2 | CA II | Proximální tubulus |
| Renální glykosurie | – | Inhibitory SGLT2sa (Canagliflozin, Tofogliflozin, Dapagliflozin) | SLC5A2 | Kotransportér sodíku a glukózy 2 (SGLT2) | Proximální tubulus |
| Bartterův syndrom | ↓ | Furosemid | SLC12A1b | Na-K-2Cl kotransportér (NKCC2) | Henleho klička |
| Gitelmanův syndrom | ↓ | Thiazid | SLC12A3 | Thiazid-citlivý Na-Cl kotransportér | Distální tubulus |
| Diabetes insipidus | – | Vaptan | AVPR2, AQP2 | V2 receptor | Sběrný kanálek |
| Pseudohypoaldosteronismus, AD | ← | Spironolakton | NR3C2 | Spironolakton-senzitivní mineralokortikoidní receptor | Sběrný kanálek |
| Pseudohypoaldosteronismus, AR | Amilorid | SCNN1A, SCNN1B, SCNN1G | Amilorid-senzitivní epiteliální sodíkový kanál | Sběrný kanálek | |
AR – autozomálně recesivní; AD – autozomálně dominantní.
Inhibitory SGLT2 (glukuretika) jsou novými léky pro léčbu diabetu 2. typu; při léčbě jsou pozorovány příhody spojené s osmotickou diurézou, jako je polakisurie a polyurie.
Přestože existují různé Bartterovy geny, tento napodobuje furosemid.
Přístup k hypokalémii
GS se také nazývá familiární hypokalemická hypomagnezémie, protože hypokalémie je nejčastějším jevem. U pacienta s hypokalémií a podezřením na tubulopatii je třeba potvrdit hyperkaliurii. Renální ztrátu kalia lze prokázat výpočtem TKKG, FeK, náhodným poměrem K/Cr nebo po 24hodinovém sběru moči (tabulka 1). Posouzení kaliurie by mělo být provedeno, pokud pacient neužívá diuretika ani suplementaci draslíku a pokud je hladina draslíku nízká. Pokud je vylučování draslíku nižší než 30 mmol/l a TTKG je nízká, je hypokalémie způsobena extrarenálními ztrátami nebo transcelulárním posunem K+.
Pokud se u pacienta s hypokalémií potvrdí nadměrné renální ztráty draslíku, je další diagnostika založena na pH a krevním tlaku. V mnoha případech tato jednoduchá vyšetření k diagnóze postačí. Důležitým problémem, o který je stále větší zájem, je primární aldosteronismus (PA). Některé klinické studie naznačují, že PA je příčinou více než 10 % arteriální hypertenze (AH) a je častější u pacientů s AH rezistentních na antihypertenziva.19 Hypokalémie je jedním z „klasických“ příznaků PA, ale není tak častá, jak se předpokládalo.20 Přesto, protože v diagnostice AP neexistuje zlatý standard, je vhodné pamatovat na osu RAA u pacientů s AH a hypokalémií.19
Diagnostika hypomagnezémie
Hypomagnezémie je častou abnormalitou u pacientů s GS, i když není pozorována ve všech případech.21 U normomagnezémických pacientů s GS jsou klinické projevy a elektrolytové abnormality mírnější.22
Podobně jako draselné ionty jsou i ty hořečnaté volně filtrovány glomeruly. V proximálním tubulu se vstřebává 10 % filtrovaného Mg2+, ve vzestupném raménku Henleovy kličky 50-70 %. Distální reabsorpce závisí na epiteliálních Mg2+ TRPM6 kanálech. Magnéziurie závisí na perorálním příjmu a ke stanovení renálního úbytku Mg2+ je třeba vypočítat FeMg (FeMg=(magnézium v moči × kreatinin v séru)/×100). FeMg nižší než 2 % svědčí o nedostatečném příjmu, gastrointestinálních ztrátách nebo přesunu Mg do buněk. FeMg nad 4 % svědčí o renální ztrátě Mg2+. Pokud je prokázán renální úbytek Mg, měl by se vypočítat náhodný poměr Ca/Cr. Poměr Ca/Cr v moči 0,3 je typický pro hypermagezurii u Bartterova syndromu, stejně tak izolovaná recesivní hypomagnezémie s normokalciurií, familiární hypomagnezémie s hyperkalciurií a nefrokalcinózou, autozomálně dominantní hypokalcemie s hyperkalciurií, léčba kličkovými diuretiky a nefropatie způsobená některými nefrotoxiny.23
Homeostáza hořčíku a draslíku spolu souvisí a depleci draslíku nelze upravit, dokud nedojde ke korekci hypomagnezémie.23 V průměru by pacienti měli dostávat až 500mEq draslíku, 4-5mg/kg/den 5-10mg chloridu hořečnatého. Užitečné jsou amilorid (5-10mg/den) a spironolakton (200-300mg).3
K diagnostice jsou nezbytné klinické příznaky a laboratorní výsledky. V případech s typickým fenotypem někteří autoři doporučují k potvrzení diagnózy provést thiazidový test.24 V prezentovaném případě genetické vyšetření odhalilo dvě nové mutace genu SLC12A3. Jejich příčinou jsou pravděpodobně chybějící funkční mutace NCCT.
Závěry
GS syndrom je jednou ze vzácných příčin hypokalémie a pro lékaře se jeví jako výzva. V tomto článku ukazujeme, že pokud si člověk vzpomene na velmi jednoduchý přístup k hypokalémii a je si vědom účinku diuretik a jejich podobnosti s dědičnými tubulopatiemi, může být diagnóza poměrně jednoduchá. GS je třeba odlišit od jiných tubulopatií (dědičných i získaných) a jiných příčin hypokalémie (např. Connovy choroby). Rodinná anamnéza může odhalit asymptomatické pacienty s GS. Vhodná léčba chrání pacienty před potenciálně nebezpečnými komplikacemi.