
Eine kurze Einführung in die ChIP-Seq

Protein-DNA-Wechselwirkungen werden in großem Umfang genutzt, um die der Zellphysiologie zugrunde liegenden Mechanismen zu entschlüsseln. Die Entwicklung der Technologie der Chromatin-Immunpräzipitation (ChIP) ermöglichte die Untersuchung solcher Mechanismen. Nach einer weiteren Entwicklung entstand eine alternative Methode für die Tiefen-Sequenzierung (ChiP-Seq), die Vorteile in Bezug auf Spezifität und Empfindlichkeit bietet.
Ein ChIP-Seq-Experiment beginnt mit einer Vernetzung der ganzen Zelle mit Formaldehyd, gefolgt von einer Sonikation und DNA-Isolierung. Danach wird die Immunpräzipitation des DNA-Protein-Komplexes durchgeführt, die aus Antikörpern besteht, die an spezifische Proteine binden. Die gebildeten Immunkomplexe werden ausgefällt und gereinigt. Schließlich wird die DNA sequenziert, wodurch hochauflösende Daten über angereicherte Stellen gewonnen werden. Mit diesem Ansatz und einer gut etablierten ChIP-Seq-Pipeline können Forscher DNA-Transkriptionsfaktoren, Stellen mit Histonmodifikationen, epigenetische Veränderungen und Signaturen von Genregulationsnetzwerken erfassen.
Klinische Relevanz und Anwendungen
Epigenetische Ungleichgewichte bei Krankheiten und Gesundheitszuständen können Histonmodifikationen und veränderte Transkriptionsfaktoren beinhalten. In diesem Zusammenhang wurden ChIP-Seq-Studien eingesetzt, um die pathologischen molekularen Mechanismen aufzuklären, die Krebs und anderen Krankheiten zugrunde liegen. ChIP-Seq-Analysen tragen auch zum Verständnis der Rolle von Transkriptionsfaktoren bei Krankheiten bei. Tatsächlich scheinen einige Transkripte während der Manifestation des klinischen Phänotyps verändert zu sein.
Überblick über die ChIP-Seq-Pipeline
Die ChIP-Seq-Analysepipeline ist die Hauptkomponente von DNA-Protein-Interaktionsprojekten und besteht aus mehreren Schritten, darunter die Verarbeitung der Rohdaten, die Qualitätskontrollanalyse, die Ausrichtung am Referenzgenom, die Qualitätsprüfung der ausgerichteten Reads, das Peak-Calling, die Annotation und die Visualisierung. Ein durchdachter Versuchsplan ist jedoch entscheidend, um qualitativ hochwertige Ergebnisse bei einem ChIP-seq-Experiment zu erhalten. Bevor Sie mit der Analyse beginnen, sollten Sie unbedingt Parameter wie Probenwiederholungen, Kontrollgruppen, Sequenzierkits und Sequenzierplattformen berücksichtigen.
Qualitätskontrolle
Alle Basepair-Berichte enthalten Qualitätsbewertungen, um potenzielle Sequenzierprobleme oder Verunreinigungen in Ihren Eingabedaten aufzudecken.
Der Schritt der Qualitätskontrolle (QC) zielt darauf ab, die Qualität von Hochdurchsatzdaten zu bewerten, die durch Sequenzierung erzeugt wurden. Dieser Schritt ähnelt den Schritten, die bei DNA-seq- und RNA-seq-Analysen durchgeführt werden. Hier werden hauptsächlich die Sequenz- und Basenqualität, der GC-Gehalt, das Vorhandensein von Sequenzieradaptern und überrepräsentierte Sequenzen bewertet. Eines der am häufigsten verwendeten Programme für diese Art der Analyse ist FastQC. Wenn Sequenzen von geringer Qualität identifiziert werden, können sie später während des Trimming-Schrittes entfernt werden. Obwohl es sich um einen optionalen Schritt handelt, verbessert das Trimming die Datenqualität, da nur qualitativ hochwertige Reads erhalten bleiben.
Alignment
Nach der QC-Messung werden die ChIP-Seq-Reads an ein Referenzgenom angeglichen. Das Read-Mapping ermöglicht es den Forschern, den Ursprung einer Lesesequenz im Genom zu identifizieren. Zu den gängigen Alignment-Softwaretools gehören Bowtie und BWA, die beide in den ChIP-Seq-Pipelines von Basepair verwendet werden. Beide Tools bilden gering abweichende Sequenzen gegen ein Referenzgenom ab.
Der Read-Count-Fluss hilft dabei, einen Überblick über die verwertbaren Reads am Ende der Trimming-, Alignment- und Deduplizierungsprozesse zu erhalten. Stellen Sie sich die Abbildung wie ein Fließband der Datenanalyse vor: Eingabe der Rohdaten, Ausgabe der verwertbaren Reads.
Qualitätsprüfung der ausgerichteten Reads
Der nächste Schritt besteht in der Qualitätskontrolle des ausgerichteten Datensatzes. Während des Mapping-Prozesses verursachen Leseduplikate, die durch PCR-Amplifikation und Sequenzierung eingeführt werden, Verzerrungen beim Peak-Calling und der Anreicherungsanalyse. Basepair verwendet das Picard-Tool, um Duplikate zu entfernen. Sobald die Duplikate entfernt wurden, sollten Sie die Non-Redundant Fraction (NRF) der ausgerichteten Reads auswerten. NRF misst die eindeutigen Reads, die auf das Referenzgenom abgebildet werden. Ideale ChIP-seq-Experimente sollten weniger als drei Reads pro Position aufweisen.
Peak Calling
Der Peak-Calling-Schritt erkennt angereicherte Protein-DNA-Interaktionsregionen im Genom. Die ChIP-seq-Pipeline von Basepair verwendet MACS2 zur Durchführung dieser Analyse. In MACS2 wird das Peak-Calling in drei Hauptschritten durchgeführt: Fragmentschätzung, gefolgt von der Identifizierung lokaler Rauschparameter und schließlich der Peak-Identifizierung. Als Ergebnis dieses Schritts erhalten die Benutzer eine endgültige Tabelle mit Peak-Informationen, wie Anreicherungs-Score, -log10p-Wert, -log10q-Wert und die Position zum Peak-Start. Die Verwendung von Kontrollproben wird in diesem Schritt zum Vergleich mit dem untersuchten Zieldatensatz dringend empfohlen. Beachten Sie, dass gute Kontrollgruppen zuverlässigere Ergebnisse liefern.
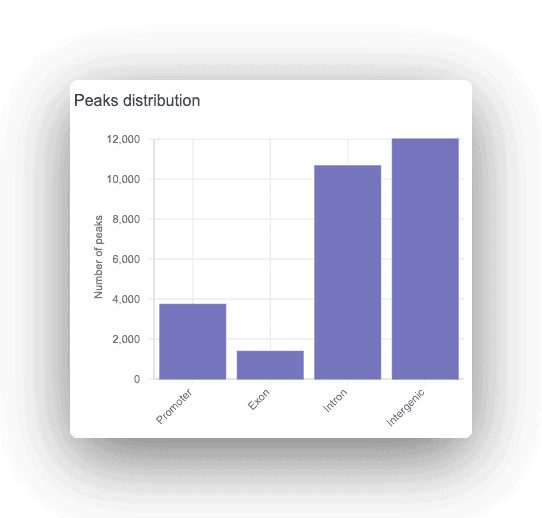
Jeder Peak wird als Promotor, intronisch oder intergenisch annotiert, wobei das entsprechende Gen angezeigt wird. Für alle gefundenen Peaks wird eine Motivanalyse durchgeführt, um überrepräsentierte Transkriptionsfaktor-Bindungsstellen zu finden.
Überblick über die Ergebnisse
Eine ChIP-seq-Pipeline kann nicht nur Informationen über den Chromatinzustand liefern, sondern auch über die Bindung von Transkriptionsfaktoren in einem bestimmten Gen oder Loci-Kontext. Das Auftreten von Histonmodifikationen und Transkriptionsfaktoren in DNA-Regulationsregionen kann eine zustandsspezifische epigenetische Signatur darstellen. So können epigenetische Störungen mit klinischen Phänotypen in Verbindung gebracht werden. So kann beispielsweise die Heterogenität des Chromatinzustands zu einer Behandlungsresistenz bei Brustkrebs führen. Diese Zellen neigen dazu, repressive Histonmodifikationsmarker zu verlieren und die Expression von Genen, die bekanntermaßen die Resistenz gegen eine Krebsbehandlung fördern, weiter zu erhöhen.
Peak-, Motiv- und Pathway-Analyse in der ChIP-Seq-Analyse-Pipeline
Die Identifizierung der Motivanreicherung von Transkriptionsfaktoren wird verwendet, um zu klären, ob Transkriptionsfaktoren in einer bestimmten Region kooperieren oder konkurrieren. Die Identifizierung von Peaks in DNA-Motivregionen kann die Interpretation der experimentellen Ergebnisse verbessern. Sowohl die Peak- als auch die Motivanalyse geben Aufschluss darüber, was in einer Zelle vor sich geht. Die Integration von Peak- und Motivanreicherungen führt zu einer epigenomischen Landschaft mit möglichen biologischen Konsequenzen. Darüber hinaus wird die Analyse von Signalwegen genutzt, um Proteine in einem Signalweg zu identifizieren. Untersuchungen und Schlussfolgerungen werden auf der Grundlage der Anwesenheit von Proteinen formuliert.
Datenvisualisierung
Die aus einer ChIP-seq-Pipeline resultierenden Daten können mit einem Genombrowser visualisiert werden. Basepair-Berichte enthalten einen eingebetteten IGV2-Genombrowser, mit dem Sie mit Ihren Daten interagieren können. Die Daten können alternativ mit Heatmaps visualisiert werden, d. h. mit repräsentativen, auf der Datendichte basierenden Intensitätsinfografiken, die das Vorhandensein oder Fehlen bestimmter Markierungen anzeigen. Andere hier verwendete Grafiken sind Anreicherungsdiagramme, upSet und Abdeckungsdiagramme, die die Abdeckung von Peak-Regionen im Genom berechnen und anzeigen.
Der Genom-Browser ist ein hervorragendes Werkzeug zur Visualisierung Ihrer genomischen Rohdaten. Er ist in jeden ChIP-seq-Analysebericht auf Basepair integriert.
1. Grosselin, K., A. Durand, et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet, v.51, n.6, Jun, p.1060-1066. 2019.
2. Northrup, D. L. e K. Zhao. Anwendung von ChIP-Seq und verwandten Techniken zur Untersuchung der Immunfunktion. Immunity, v.34, n.6, Jun 24, p.830-42. 2011.
3. Park, S. J., J. H. Kim, et al. A ChIP-Seq Data Analysis Pipeline Based on Bioconductor Packages. Genomics Inform, v.15, n.1, Mar, p.11-18. 2017.
4. Pepke, S., B. Wold, et al. Computation for ChIP-seq and RNA-seq studies. Nat Methods, v.6, n.11 Suppl, Nov, p.S22-32. 2009.
5. Satoh, J., N. Kawana, et al. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul Syst Bio, v.7, p.139-52. 2013.