
Einführung
Hypokaliämie ist ein häufiges klinisches Problem in der Praxis von Endokrinologen und Nephrologen. Es gibt viele offensichtliche Ursachen für Hypokaliämie wie Durchfall, Erbrechen oder Diuretika-Missbrauch. Andere Ursachen, wie z. B. Tubulopathien, werden nur selten beobachtet und ihre Diagnose ist schwieriger. Es gibt viele vererbte und erworbene Tubulopathien, die eine Hypokaliämie verursachen, die manchmal schwerwiegend und lebensbedrohlich ist.1
Eine relativ häufige, aber übersehene Ursache für Hypokaliämie ist das Gitelman-Syndrom (GS).2 Es handelt sich um eine rezessive salzverlierende Tubulopathie, die durch eine Mutation des SLC12A3-Gens verursacht wird. Das SLC12A3-Gen kodiert für den thiazidempfindlichen Transporter NCCT (Natriumchlorid-Co-Transporter). NCCT befindet sich in den distalen tubulären Faltenzellen (DCC), die für 7-10 % der tubulären Elektrolytresorption verantwortlich sind.3
Die schwerwiegendsten Laboranomalien, die bei GS gefunden werden, sind Hypokaliämie und Hypomagnesiämie, die durch renalen K+- und Mg2+-Verlust verursacht werden. Weitere typische Veränderungen sind metabolische Alkalose, Hypokalziurie und hyperreninämischer Hyperaldosteronismus.4 Häufig wird eine leichte bis mäßige Hypophosphatämie beobachtet.5 Auch über eine schwere Hypophosphatämie mit schwerer Hyponatriämie wurde berichtet.6,7
Die ersten Symptome von GS treten bei Kindern oder jungen Erwachsenen mit normalem Wachstum und salzsüchtigem Verhalten auf (Kinder, die eifrig Essiggurken oder Salzlake, gesalzene Gurken, Orangen und Zitronen verzehren, Kinder, die Salz von Kartoffelchips lecken usw.).8 Die klinische Präsentation variiert von Patient zu Patient. Einige sind asymptomatisch, andere entwickeln lebensbedrohliche Komplikationen. Männer zeigen einen schwereren Phänotyp als Frauen.8 Die häufigsten Symptome sind Muskelkrämpfe und -schwäche, Verstopfung, Nykturie, Polyurie, Durst, Polydipsie, Herzrhythmusstörungen, Parästhesien und erhöhter Salzappetit. Arterielle Hypotonie ist häufig und in vielen Fällen das auffälligste Symptom, allerdings kann in der alternden GS-Population auch Bluthochdruck auftreten.8 Die Korrelation zwischen biochemischen Anomalien und Symptomen ist nicht sehr ausgeprägt.9 GS beeinträchtigt die Stimmung und die sozialen Beziehungen von Kindern nicht.9,10 Ansonsten sind die Symptome bei Erwachsenen häufiger und können sich negativ auf ihre Lebensqualität auswirken. Fünfundvierzig Prozent der GS-Patienten betrachten ihre Symptome als mäßiges bis großes Problem.11 Extreme Erschöpfung, Muskelschwäche, Parästhesien, starke Müdigkeit und Hypotonie sind mit einer leichten bis starken Einschränkung der täglichen Aktivitäten verbunden.9
Die geschätzte Prävalenz von GS liegt bei 1:40.0008 und die Prävalenz von heterozygoten Mutationen beträgt mindestens 1 % in der europäischen Bevölkerung. Bisher wurden mehr als 180 verschiedene Mutationen in SLC12A3 beschrieben.12
Fallbericht
Ein 26-jähriger Mann wurde aufgrund von Synkopen, generalisierter Muskelschwäche und Muskelkrämpfen ins Krankenhaus eingeliefert. Die Anamnese des Patienten ergab eine Synkope mit einem Kaliumspiegel von 3,16 mmol/l. Bei der weiteren ambulanten Nachuntersuchung wurde ein wiederholtes Auftreten von Hypokaliämie (niedrigster Wert 2,6 mmol/l) festgestellt. Der Blutdruck war normal 110/80, die Herzfrequenz betrug 72 pro Minute; bei der körperlichen Untersuchung gab es keine Veränderungen. Kein neurologischer Befund, Gewicht 74 kg, Größe 178 cm. Bei der Aufnahme ins Krankenhaus ergab die Laboruntersuchung eine leichte Hypokaliämie (K+ 3,0mmol/l), Hypomagnesiämie (Mg2+ 1,36mg/dl), Hypocalciurie (
mg/24h) und metabolische Alkalose (HCO3- 29,7mmol/l, BE 5,3mmol/l). Die Nierenfunktion war mit einer eGFR>60ml/min gut. Die bildgebenden Untersuchungen waren unauffällig, ebenso das EKG.
Weitere Untersuchungen bestätigten, dass die Hypokaliämie durch einen renalen Kaliumverlust verursacht wurde. Der 24-Stunden-Kaliumverlust, der transtubuläre K+-Gradient (TTKG), die fraktionierte K+-Ausscheidung (FeK) und das zufällige K/Kreatinin-Verhältnis (K/Cr) waren typisch für eine Hyperkaliurie und sind in Tabelle 1 aufgeführt.13-15 Die tägliche Magnesiurie betrug 64mg/24h, die fraktionierte Magnesiumausscheidung (FeMg) 15%.
Die diagnostischen Werte für Hypokaliämie.13-15
| Diagnosewerte für unangemessene Kaliurese | Patient | |
|---|---|---|
| Harn-K+ | >20mmol/24h Wenn Serum-K+3.5mmol/l |
57mmol/l (Serum K+=3.0mmol/l) |
| FeK=×100% | >6.5% Wenn Serum K+3.5mmol/l |
12% (Serum K+=3.0mmol/l) |
| TTKG= | TTKG>2 Wenn Serum K+3.5mmol/l |
7.3 (Serum K+=3.0mmol/l) |
| uK/Cr – zufälliges Urin K+ zu Kreatinin Verhältnis | uK/Cr>15mmol/g Wenn Serum K+3.5mmol/l |
67mmol/g (Serum K+=3.0mmol/l) |
Obwohl der Blutdruck normal war, wurden hormonelle Tests zum Ausschluss des Conn’schen Syndroms und ein CT des Abdomens durchgeführt, die keine Anomalien der Nebennieren ergaben. Ein sekundärer Hyperaldosteronismus mit Aldosteronspiegeln von 289pg/ml (Normalbereich 20-180) und Reninspiegeln von 205mIU/ml (Normalbereich 2,8-39,9) war typisch für GS.
Der Patient wurde aufgrund des klinischen Phänotyps als GS diagnostiziert. Nach der Diagnose wurde eine Behandlung mit Kalium- und Magnesiumzusätzen eingeleitet. Er erhielt 20 mmol Kaliumchlorid und 18 mmol Magnesiumpyrrolidoncarboxylat, was zu einer klinischen Verbesserung führte. Die Familienanamnese ergab eine leichte asymptomatische Hypokaliämie bei der 35-jährigen Schwester des Patienten. Die Untersuchungen ergaben jedoch weder eine Hyperkaliurie noch eine Hypomagnesiämie. GS wurde in diesem Fall nicht diagnostiziert.
Genetische Analyse
Nach der genetischen Analyse, dem von Nozu16 vorgeschlagenen Algorithmus, sollte das SLC12A3-Gen bei Patienten mit hypokaliämischer metabolischer Alkalose, mit Vollgeburt, Normalgewicht ohne Nephrokalzinose, mit Hypokalzurie und Hypomagnesiämie getestet werden.
In dem beschriebenen Fall wurden genetische Tests durchgeführt und zwei heterozygote Mutationen festgestellt: c.35_36insA und c.1095+5G>A wurden im Transkript NM_000339.2 des SLC12A3-Gens gefunden (Abb. 1). Beide Mutationen sind noch nicht in der HGMD(r)-Datenbankversion verfügbar. Die erste Mutation wurde auch bei der Mutter des Patienten gefunden, die zweite beim Vater. Nur eine der beiden bei unserem Patienten identifizierten Mutationen c.35_36insA wurde bei seiner Schwester gefunden.
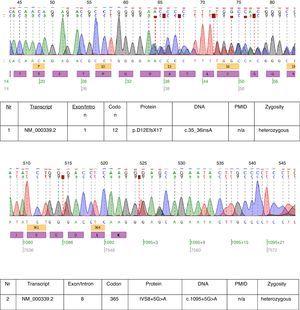
Chromatogramme der DNA-Sequenzierung des betroffenen Patienten.
Die bei diesem Patienten gefundenen Mutationen belegen nahezu die klinische Diagnose von GS. Die pathogenetische Bedeutung der Frameshift-Mutation ist offensichtlich. Die pathogenetische Relevanz der Spleißstellenmutation von Intron 8 ist nicht sicher. Die Konsensus-Spleißsequenz G (100%), T (100%), A (62%), A (68%), G (82%), T (63%) am Spleißspender von Intron 8 stimmt nicht genau mit dem Konsensus GTgAGc überein. Die bei diesem Patienten gefundene Mutation weicht noch weiter von der Konsenssequenz ab (GtgAac). Auch die In-silico-Analyse durch Mutationstester hält diese Mutation für krankheitsverursachend.17
Diskussion
Tubulopathien sind seltene Krankheiten. Laut der RenalTube-Datenbank18 sind die häufigsten primären Tubulopathien in der europäischen Bevölkerung die distale renale tubuläre Azidose, das Bartter-Syndrom, die familiäre Hypomagnesiämie mit Hyperkalziurie und die GS.18 Die Prävalenz der GS liegt bei etwa 25 Fällen pro 1 Million Menschen.3 Da die GS eine der häufigsten ist, werden wahrscheinlich viele Nephrologen und Endokrinologen im Laufe ihrer Karriere mit Fällen von GS konfrontiert. Unser Patient hatte das typische klinische Bild einer GS und sprach gut auf die Therapie an. Das Problem bei GS ist, dass eine übersehene Hypokaliämie zum Tod durch Herzstillstand oder Lähmung der Atemmuskulatur führen kann. Schwere neuromuskuläre Symptome wie hypokaliämische Lähmungen treten bei bis zu 6 % der Patienten auf (häufiger bei asiatischen Patienten).3
Die vererbten Tubulopathien haben eine Art „Spiegelbild“ – erworbene Tubulopathien, die durch Diuretika verursacht werden. Jedes Diuretikum (genauer gesagt: Natriuretika, Acaretika oder Glucuretika) verursacht Anomalien, die denen der vererbten Tubulopathien ähnlich sind (Tabelle 2). Fast bei jeder Tubulopathie kommt es zu einer Polyurie, und Veränderungen des Kaliumspiegels gehören zu den häufigsten Problemen. Bei Patienten mit GS werden Mutationen im SLC12A3-Gen gefunden. Dieses Gen kodiert den Thiazid-sensitiven Transporter. Daher ähnelt GS einer chronischen Behandlung mit Thiaziden.
Diuretika und Tubulopathien.
| Ererbter einzelner Gendefekt | Kaliumkonzentration | Symptome ähnlich der Verwendung von: | Gen | Genprodukt | Genproduktstandort |
|---|---|---|---|---|---|
| Guibaud-Vainsel-Syndrom (RTA 3) | ↓ | Acetazolamid | CA2 | CA II | Proximaler Tubulus |
| Renale Glykosurie | – | SGLT2-Inhibitorena (Canagliflozin, Tofogliflozin, Dapagliflozin) | SLC5A2 | Natrium/Glucose-Cotransporter 2 (SGLT2) | Proximaler Tubulus |
| Bartter-Syndrom | ↓ | Furosemid | SLC12A1b | Na-K-2Cl-Cotransporter (NKCC2) | Henle-Schleife |
| Gitelman-Syndrom | ↓ | Thiazid | SLC12A3 | Thiazid-sensitiver Na-Cl-Cotransporter | Distaler Tubulus |
| Diabetes insipidus | – | Vaptan | AVPR2, AQP2 | V2-Rezeptor | Sammelgang |
| Pseudohypoaldosteronizm, AD | ← | Spironolacton | NR3C2 | Spironolacton-empfindlicher Mineralocorticoid-Rezeptor | Sammelkanal |
| Pseudohypoaldosteronizm, AR | Amilorid | SCNN1A, SCNN1B, SCNN1G | Amilorid-empfindlicher epithelialer Natriumkanal | Sammelkanal | |
AR – autosomal rezessiv; AD – autosomal dominant.
SGLT2-Hemmer (Glucuretika) sind die neuen Medikamente zur Behandlung des Typ-2-Diabetes; während der Behandlung werden osmotische Diurese-bedingte Ereignisse wie Pollakiurie und Polyurie beobachtet.
Obwohl es verschiedene Bartter-Gene gibt, ist dies dasjenige, das Furosemid imitiert.
Hypokaliämie
GS wird auch als familiäre hypokaliämische Hypomagnesiämie bezeichnet, weil Hypokaliämie das häufigste Phänomen ist. Bei einem Patienten mit Hypokaliämie und Verdacht auf Tubulopathie sollte man eine Hyperkaliurie bestätigen. Ein renaler Kaliumverlust kann durch die Berechnung des TKKG, des FeK, des zufälligen K/Cr-Verhältnisses oder nach einer 24-Stunden-Urinsammlung nachgewiesen werden (Tabelle 1). Die Beurteilung der Kaliurie sollte durchgeführt werden, wenn der Patient weder Diuretika noch eine Kaliumsupplementierung einnimmt und wenn der Kaliumspiegel niedrig ist. Wenn die Kaliumausscheidung unter 30 mmol/l liegt und der TTKG-Wert niedrig ist, wird die Hypokaliämie durch einen extrarenalen Verlust oder eine transzelluläre K+-Verschiebung verursacht.
Wenn ein übermäßiger renaler Kaliumverlust bei einem Patienten mit Hypokaliämie bestätigt wird, basiert die weitere Diagnose auf pH-Wert und Blutdruck. In vielen Fällen reichen diese einfachen Untersuchungen für die Diagnose aus. Ein wichtiges Problem, das zunehmend an Bedeutung gewinnt, ist der primäre Aldosteronismus (PA). Einige klinische Studien legen nahe, dass PA die Ursache für mehr als 10 % der arteriellen Hypertonie (AH) ist und häufiger bei Patienten mit AH auftritt, die gegen blutdrucksenkende Mittel resistent sind.19 Hypokaliämie ist eines der „klassischen“ Symptome von PA, kommt aber nicht so häufig vor, wie angenommen wurde.20 Da es jedoch keinen Goldstandard für die Diagnose von AP gibt, sollte man bei Patienten mit AH und Hypokaliämie an die RAA-Achse denken.19
Diagnose der Hypomagnesiämie
Die Hypomagnesiämie ist eine häufige Anomalie bei GS-Patienten, obwohl sie nicht in jedem Fall beobachtet wird.21 Bei normomagnesiämischen Patienten mit GS sind die klinischen Manifestationen und Elektrolytanomalien milder.22
Gleich wie die Kaliumionen werden auch die Magnesiumionen von den Glomeruli frei gefiltert. 10 % des gefilterten Mg2+ wird vom proximalen Tubulus absorbiert, 50-70 % im aufsteigenden Teil der Henle-Schleife. Die distale Rückresorption hängt von den epithelialen Mg2+ TRPM6-Kanälen ab. Eine Magnesiurie hängt von der oralen Aufnahme ab, und das FeMg sollte berechnet werden, um einen renalen Mg2+-Verlust festzustellen (FeMg=(Magnesium im Urin×Kreatinin im Serum)/×100). FeMg unter 2 % deutet auf eine schlechte Aufnahme, GI-Verluste oder eine Verlagerung von Mg in die Zellen hin. FeMg über 4 % deutet auf einen renalen Mg2+-Verlust hin. Wenn ein renaler Mg-Verlust nachgewiesen wird, sollte das Ca/Cr-Verhältnis stichprobenartig berechnet werden. Ein Ca/Cr-Verhältnis von 0,3 im Urin ist typisch für eine Hypermagesurie beim Bartter-Syndrom, ebenso wie eine isolierte rezessive Hypomagnesiämie mit Normocalciurie, eine familiäre Hypomagnesiämie mit Hypercalciurie und Nephrocalcinose, eine autosomal-dominante Hypocalciämie mit Hypercalciurie, eine Schleifendiuretika-Behandlung und eine durch einige Nephrotoxine verursachte Nephropathie.23
Magnesium- und Kaliumhomöostase sind miteinander verbunden, und eine Kaliumdepletion kann erst korrigiert werden, wenn die Hypomagnesiämie behoben ist.23 Im Durchschnitt sollten die Patienten bis zu 500mEq Kalium und 4-5mg/kg/Tag 5-10mg Magnesiumchlorid erhalten. Amilorid (5-10mg/Tag) und Spironolacton (200-300mg) sind hilfreich.3
Klinische Symptome und Laborergebnisse sind für die Diagnose entscheidend. In Fällen mit typischem Phänotyp empfehlen einige Autoren die Durchführung eines Thiazid-Tests zur Bestätigung der Diagnose.24 Im vorliegenden Fall ergab die genetische Untersuchung zwei neue Mutationen des SLC12A3-Gens. Sie werden wahrscheinlich durch fehlende funktionelle Mutationen des NCCT verursacht.
Schlussfolgerungen
Das GSS-Syndrom ist eine der seltenen Ursachen für Hypokaliämie und stellt für Ärzte eine Herausforderung dar. Wir zeigen in diesem Beitrag, dass die Diagnose recht einfach sein kann, wenn man sich an einen sehr einfachen Ansatz zur Hypokaliämie erinnert und sich der Wirkung von Diuretika und ihrer Ähnlichkeit mit vererbten Tubulopathien bewusst ist. GS sollte von anderen Tubulopathien (sowohl vererbt als auch erworben) und anderen Ursachen für Hypokaliämie (z. B. Morbus Conn) unterschieden werden. Die Familienanamnese kann asymptomatische Patienten mit GS aufdecken. Eine geeignete Behandlung schützt die Patienten vor potenziell gefährlichen Komplikationen.