
Anmerkung des Herausgebers: Platin-Highlight-Artikel sind bemerkenswerte Veröffentlichungen, die in regelmäßigen Abständen von Dr. Craig Reynolds, stellvertretender Direktor des National Cancer Institute, aus den zuletzt veröffentlichten Platin-Publikationen ausgewählt werden.![]()
Als Alison Rattray und ihre Kollegen vom Gene Regulation and Chromosome Biology Laboratory (GRCBL) eine mutierte Hefezelle untersuchten, die sie in einem Screening isoliert hatten, fiel ihnen etwas Seltsames auf.
Die DNA wies eine „sehr spezifische, aber seltsame Umstrukturierung“ auf, erklärte sie. Die Anordnung stellte sich als DNA-Palindrom heraus und „öffnete die Tür zur Untersuchung dieser schwer fassbaren DNA-Motive“, sagte sie.
Eine wissenschaftliche Mitarbeiterin des GRCBL, NCI Center for Cancer Research, sagte, dass ihre Entdeckung zwar schon einige Jahre zurückliegt, ihre Gruppe aber weiterhin DNA-Reparaturen und Umlagerungen untersucht, die zu abnormalen Reparaturen führen, „weil sie mit bestimmten Krebsarten in Verbindung stehen“. Das Ziel der Gruppe ist es, den Ursprung dieser Rearrangements besser zu verstehen, fügte sie hinzu.
Rattray ist einer der Hauptautoren der in BMC Genomics veröffentlichten Forschungsarbeit, die die von der Gruppe neu entwickelte Methode zur genomweiten Sequenzierung von DNA-Palindromen in einer Krebszelllinie beschreibt.
Was ist ein DNA-Palindrom?
Eine palindromische Sequenz von Nukleotiden (die mit A, T, C oder G gekennzeichnet sind) liegt vor, wenn komplementäre DNA-Stränge in beiden Richtungen gleich gelesen werden, entweder vom 5-Prime-Ende oder vom 3-Prime-Ende. Zum Beispiel wird die Sequenz GGATCC auf einem DNA-Strang als Palindrom betrachtet, weil die Sequenz auf dem komplementären Strang CCTAGG ist.
Aufgrund der Sequenzkomplementarität können sich palindromische Sequenzen auch auf sich selbst zurückfalten und Haarnadelschleifen oder Kreuzungen bilden, die aus der normalen Doppelhelix herausragen, so Rattray. „Kleine Haarnadeln sind nicht problematisch, aber wenn Palindrome lang sind (mehr als 100 Basenpaare), stören sie normale zelluläre Prozesse wie Transkription und Replikation“, erklärte sie.
Einige Krebszellen weisen massive Genomumlagerungen auf, zu denen Genamplifikationen, Translokationen und Deletionen gehören, und diese Umlagerungen sind oft mit dem Vorhandensein eines Palindroms verbunden, was auf einen möglichen Zusammenhang zwischen dem Palindrom und den Genumlagerungen hindeutet. Wie diese Ereignisse zustande kommen, ist nicht genau bekannt. Was jedoch bekannt ist, ist, dass solche Rearrangements mit dem Fortschreiten und der Prognose des Krebses in Verbindung stehen, so Rattray.
Neue Hypothese für genomische Rearrangements
Das bevorzugte Modell, das ursprünglich vor mehr als 60 Jahren von Barbara McClintock, Ph.D., vorgeschlagen wurde, besagt laut Rattray, dass nach einem Chromosomenbruch Schwesterchromatiden replizieren und fusionieren, wodurch ein Chromosom mit zwei Zentromeren entsteht, die durch ein DNA-Palindrom verbunden sind. Im McClintock-Modell führt das Vorhandensein von zwei Zentromeren zu weiteren genomischen Umlagerungen.
Rattray sagte jedoch, dass ihre Gruppe und andere gezeigt haben, dass „DNA-Palindrome instabil sind und von sich aus zu genomischen Umlagerungen führen können, was darauf hindeutet, dass Palindrome nicht nur durch die Fusion von Schwesterchromatiden, sondern auch durch andere Mechanismen wie Replikationsfehler entstehen können.“
Die Gruppe stellte die Hypothese auf, dass „bei Krebserkrankungen, die massive Umlagerungen erfahren, die Zellen anfällig für die Bildung von Palindromen sind, und wenn sie einmal gebildet sind, führt die Instabilität des Palindroms zu weiteren Umlagerungen, einschließlich Genamplifikationen, Translokationen und Deletionen“, sagte Rattray. „Jede Genumlagerung ist mutagen, und Umlagerungen, die das zelluläre Wachstum fördern, wie bei Krebs, werden natürlich durch Selektion begünstigt.“
Neue Technologie identifiziert und charakterisiert Palindrome
Die Forscher haben eine Technologie entwickelt, die es ihnen ermöglicht, Tumore zu untersuchen, mit dem Ziel, die Wahrscheinlichkeit der Palindrombildung in diesen Tumoren zu verstehen, so Rattray. Sie hoffen zu erfahren, welche Ereignisse solche instabilen Formationen auslösen, und dieses neue Verständnis könnte zu neuen Behandlungen führen. So habe die Gruppe bereits festgestellt, dass bestimmte Hefezellen, die für die Palindrombildung anfällig sind, viel empfindlicher als normale Zellen auf Strahlung sowie auf Verbindungen reagieren, die häufig bei der Behandlung von Krebs eingesetzt werden, wie z. B. Cisplatin.
„Derzeit versuche ich, Methoden zur selektiven Anreicherung von Palindromen aus dem Rest der zellulären DNA zu etablieren, was eine größere Empfindlichkeit bei der Analyse des Palindromgehalts von Krebszellen ermöglichen wird“, sagte sie. Bei der bisherigen Methode verloren die Forscher die Kreuzungssequenzen, die Hinweise auf den Ursprung der Palindrome geben könnten, und mussten sie einzeln analysieren, erklärte sie. „Wir haben jetzt gezeigt, dass die PacBio-Plattform ohne weiteres ein DNA-Palindrom sequenzieren kann“, sagte sie.
Rattray promovierte an der University of Washington in Seattle, wo sie die Replikation von Retroviren untersuchte. Nach einem Postdoc-Stipendium an der Columbia University, wo sie DNA-Rekombination und durch DNA-Doppelstrangbrüche induzierte Umlagerungen in Hefe untersuchte, kam sie zum NCI in Frederick und arbeitete im Labor von Jeffrey Strathern, Ph.D., Leiter des GRCBL.
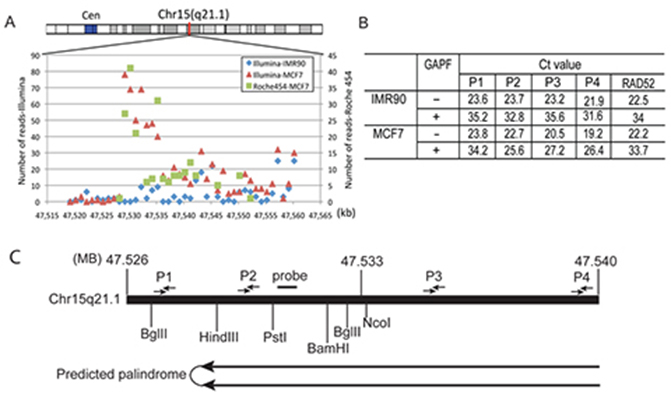
Palindrom-Kartierungsstrategie. (A) Read-Dichte-Verteilung in der Chr15q21.1: 47.529.204-47.550.373 Region, dargestellt als 1-kb-Bins. (B) qPCR-Analyse zur Überwachung der Palindrom-Anreicherung und zur Bestimmung der Richtungsabhängigkeit des Chr15q21.1-Palindroms. Die Forscher berechneten das Ausmaß der Verarmung einer bestimmten TaqMan-Primerset-Region auf der Grundlage des Ct-Werts vor und nach dem GAPF-Protokoll sowohl in IMR-90- als auch in MCF-7-Proben. Die Faltenanreicherung basiert auf dem Vergleich der Faltendepletion zwischen verschiedenen Primersätzen (P1, P2, P3 und P4) im Verhältnis zu einer Einzelkopie-Sequenz im Genom (RAD52). Die Positionen der TaqMan-Primersätze P1, P2, P3 und P4 sind in (C), Karte der genomischen Region Chr15: 47.520.000-47.550.000 mit Restriktionsstellen und Primerpositionen, angegeben. Abbildung aus Yang et al., GAP-Seq: a method for identification of DNA palindromes, BMC Genomics 2014, 15:394; doi:10.1186/1471-2164-15-394.