
Introducción
La hipocalemia es un problema clínico común en la consulta de endocrinólogos y nefrólogos. Hay muchas causas evidentes de hipopotasemia como la diarrea, los vómitos o el abuso de diuréticos. Otras causas como las tubulopatías se observan raramente y su diagnóstico es más difícil. Hay muchas tubulopatías heredadas y adquiridas que causan hipocalemia, a veces grave y potencialmente mortal.1
Una causa relativamente común pero ignorada de hipocalemia es el síndrome de Gitelman (GS).2 Es una tubulopatía perdedora de sal recesiva causada por la mutación del gen SLC12A3. El gen SLC12A3 codifica el transportador sensible a la tiazida NCCT (cotransportador de cloruro de sodio). El NCCT se localiza en las células tubulares contorneadas distales (DCC), que son responsables del 7-10% de la absorción tubular de electrolitos.3
Las anomalías de laboratorio más graves que se encuentran en la GS son la hipopotasemia y la hipomagnesemia causadas por el desgaste renal de K+ y Mg2+. Otros cambios típicos son la alcalosis metabólica, la hipocalciuria y el hiperaldosteronismo hiperreninémico.4 Con frecuencia se observa una hipofosfatemia de leve a moderada.5 También se ha informado de una hipofosfatemia grave con hiponatremia severa.6,7
Los primeros síntomas de la GS se producen en niños o adultos jóvenes con un crecimiento normal y antecedentes de comportamientos de avidez por la sal (niños ávidos de consumir encurtidos o salmueras, pepinos, naranjas y limones salados, niños que lamen la sal de las patatas fritas, etc.).8 La presentación clínica varía entre los pacientes. Algunos son asintomáticos, pero otros desarrollan complicaciones potencialmente mortales. Los varones manifiestan un fenotipo más grave que las mujeres.8 Los síntomas más comunes son calambres y debilidad muscular, estreñimiento, nicturia, poliuria, sed, polidipsia, arritmias cardíacas, parestesias y aumento del apetito por la sal. La hipotensión arterial es frecuente y, en muchos casos, el síntoma más destacado; sin embargo, en la población de edad avanzada de la GS puede producirse hipertensión.8 La correlación entre las anomalías bioquímicas y los síntomas no es fuerte.9 La GS no interfiere en el estado de ánimo ni en las relaciones sociales de los niños.9,10 Por el contrario, los síntomas son más frecuentes en los adultos y pueden tener un impacto negativo en su calidad de vida. El 45% de los pacientes con GS consideran sus síntomas como un problema de moderado a grande.11 El agotamiento extremo, la debilidad muscular, las parestesias, la fatiga severa y la hipotensión se asocian con una reducción de leve a grave de las actividades diarias.9
La prevalencia estimada de la GS es de 1:40.0008 y la prevalencia de heterocigotos es de al menos el 1% en la población europea. Hasta ahora se han descrito más de 180 mutaciones diferentes en SLC12A3.12
Informe de un caso
Un varón de 26 años ingresó en el hospital por incidencia de síncope, debilidad generalizada y muscular y calambres musculares. La historia del paciente reveló un episodio de síncope con un nivel de potasio de 3,16 mmol/l. En el seguimiento posterior en consulta externa, se observó una incidencia recurrente de hipocalemia (el valor más bajo 2,6mmol/l). La presión arterial era normal 110/80, la frecuencia cardíaca era 72 por minuto; no había cambios en el examen físico. Tampoco hallazgos neurológicos, peso 74kg, altura 178cm. Al ingresar en el hospital, la evaluación de laboratorio mostró una hipocalemia leve (K+ 3,0mmol/l), hipomagnesemia (Mg2+ 1,36mg/dl), hipocalciuria (
mg/24h) y alcalosis metabólica (HCO3- 29,7mmol/l, BE 5,3mmol/l). La función renal era buena, con un FGe>60ml/min. Los estudios de imagen no presentaban ningún problema, al igual que el ECG.
Las investigaciones posteriores confirmaron que la hipopotasemia estaba causada por el desgaste renal de potasio. La pérdida de potasio en 24 horas, el gradiente transtubular de K+ (TTKG), la excreción fraccional de K+ (FeK) y la relación aleatoria K/creatinina (K/Cr) eran típicos de la hipercaliuria y se muestran en la tabla 1.13-15 La magnesiuria diaria fue de 64mg/24h, con una excreción fraccional de magnesio (FeMg) del 15%.
Los valores diagnósticos de la hipocalemia.13-15
| Valores diagnósticos de kaliuresis inadecuada | Paciente | |
|---|---|---|
| K+ urinario | >20mmol/24h Si el K+ sérico3.5mmol/l |
57mmol/l (K+ sérico=3.0mmol/l) |
| FeK=×100% | >6,5% Si el K+3 sérico.5mmol/l |
12% (K+ sérico=3,0mmol/l) |
| TTKG= | TTKG>2 Si K+ sérico3.5mmol/l |
7,3 (K+ sérico=3,0mmol/l) |
| uK/Cr – relación K+/creatinina en orina al azar | uK/Cr>15mmol/g Si K+ sérico3.5mmol/l |
67mmol/g (K+ sérico=3.0mmol/l) |
Aunque la presión arterial era normal, se realizaron pruebas hormonales para excluir la enfermedad de Conn y el TAC abdominal, y no revelaron ninguna anomalía en las glándulas suprarrenales. El hiperaldosteronismo secundario con niveles de aldosterona 289pg/ml (rango normal 20-180) y renina 205mIU/ml (rango normal 2,8-39,9) eran típicos de la GS.
La paciente fue diagnosticada de GS sobre la base de un fenotipo clínico. Tras el diagnóstico, se instauró un tratamiento con suplementos de potasio y magnesio. Recibió 20mmol de cloruro de potasio y 18mmol de carboxilato de pirrolidona de magnesio, con mejoría clínica. Los antecedentes familiares revelaron una leve hipopotasemia asintomática en la hermana del paciente, de 35 años. Pero las investigaciones no mostraron ni hipercaliuria ni hipomagnesemia. No se diagnosticó GS en este caso.
Análisis genético
De acuerdo con el análisis genético, algoritmo propuesto por Nozu16El gen SLC12A3 debe ser analizado en pacientes con alcalosis metabólica hipocalémica, con nacimiento a término, peso normal sin nefrocalcinosis, con hipocalcemia e hipomagnesemia.
En el caso descrito se realizaron pruebas genéticas y se encontraron dos mutaciones heterocigotas: c.35_36insA y c.1095+5G>A en el transcrito NM_000339.2 del gen SLC12A3 (Fig. 1). Ambas mutaciones aún no se encuentran en la versión disponible de la base de datos HGMD(r) . La primera mutación se encontró también en la madre de la paciente y la segunda en el padre. Sólo una de las dos mutaciones identificadas en nuestro paciente c.35_36insA se encontró en su hermana.
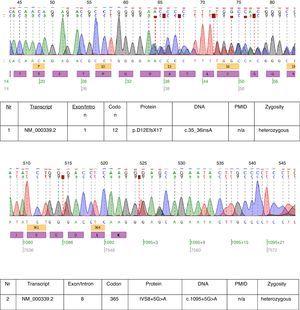
Cromatogramas de la secuenciación del ADN del paciente afectado.
Las mutaciones encontradas en este paciente casi demuestran el diagnóstico clínico de GS. La relevancia patogénica de la mutación frameshift es evidente. No podemos estar seguros de la relevancia patogénica de la mutación del sitio de empalme del intrón 8. La secuencia de empalme consensuada G (100%), T (100%), A (62%), A (68%), G (82%), T (63%) en el donante de empalme del intrón 8 no coincide exactamente con el consenso GTgAGc. La mutación encontrada en este paciente desvía aún más la secuencia del consenso (GtgAac). Además, el análisis in silico realizado por el catador de mutaciones considera esta mutación como causante de la enfermedad.17
Discusión
Las tubulopatías son enfermedades raras. Según la base de datos RenalTube18 , las tubulopatías primarias más comunes en la población europea son la acidosis tubular renal distal, el síndrome de Bartter, la hipomagnesemia familiar con hipercalciuria y la GS.18 La prevalencia de la GS es de unos 25 casos por cada millón.3 Dado que la GS es una de las más comunes, probablemente muchos nefrólogos y endocrinólogos se enfrentarán a casos de GS durante su carrera. Nuestra paciente tenía la presentación clínica típica de la GS y respondió bien al tratamiento. El problema de la GS es que la hipopotasemia que se pasa por alto puede causar la muerte por paro cardíaco o parálisis de los músculos respiratorios. El síntoma neuromuscular grave, como la parálisis hipocalémica, ocurre hasta en el 6% de los pacientes (más común en pacientes asiáticos).3
Las tubulopatías heredadas tienen una especie de «imágenes en espejo»: las tubulopatías adquiridas causadas por diuréticos. Todos los diuréticos (para ser precisos: natriuréticos, acuaréticos o glucuréticos) provocan anomalías similares a las encontradas en las tubulopatías heredadas (Tabla 2). Casi en todas las tubulopatías se produce poliuria, y las alteraciones de los niveles de potasio son uno de los problemas más comunes. En los pacientes con GS se encuentran las mutaciones del gen SLC12A3. Este gen codifica el transportador sensible a la tiazida. Por tanto, la GS se asemeja al tratamiento crónico con tiazidas.
Diuréticos y tubulopatías.
| Defecto hereditario de un solo gen | Concentración de potasio | Síntomas similares al uso de: | Geno | Producto génico | Localización del producto génico |
|---|---|---|---|---|---|
| Guibaud-Vainsel (RTA 3) | ↓ | Acetazolamida | CA2 | CA II | Túbulo proximal |
| Glicosuria renal | – | Inhibidores del SGLT2a (Canagliflozin, Tofogliflozin, Dapagliflozin) | SLC5A2 | Cotransportador de sodio/glucosa 2 (SGLT2) | Túbulo proximal |
| Síndrome de Bartter | ↓ | Furosemida | SLC12A1b | Na-K-2Cl (NKCC2) | Bucle de Henle |
| Síndrome de Gitelman | ↓ | Tiazida | SLC12A3 | Tiazida-cotransportador Na-Cl sensible | Túbulo distal |
| Diabetes insípida | – | Vaptan | AVPR2, AQP2 | Receptor V2 | Conducto colector |
| Pseudohipoaldosteronizm, AD | ← | Espironolactona | NR3C2 | Receptor mineralocorticoide sensible a la espironolactona | Ducto colector |
| Pseudohipoaldosteronizm, AR | Amilorida | SCNN1A, SCNN1B, SCNN1G | Canal de sodio epitelial sensible a la amilorida | Ducto colector | |
AR – autosómico recesivo; AD – autosómico dominante.
Los inhibidores del SGLT2 (glucuréticos) son los nuevos fármacos para el tratamiento de la diabetes de tipo 2; durante el tratamiento se observan acontecimientos relacionados con la diuresis osmótica, como la polaquiuria y la poliuria.
Aunque existen diferentes genes Bartter este es el que imita a la furosemida.
Aproximación a la hipocalemia
La SG también se conoce como hipomagnesemia hipocalémica familiar, porque la hipocalemia es el fenómeno más común. En un paciente con hipocalemia y sospecha de tubulopatía hay que confirmar la hipercaliuria. La pérdida renal de potasio puede comprobarse mediante el cálculo de la TKKG, la FeK, el cociente K/Cr aleatorio o tras la recogida de orina de 24 horas (Tabla 1). La evaluación de la caliuria debe realizarse cuando el paciente no toma diuréticos ni suplementos de potasio y cuando el nivel de potasio es bajo. Cuando la excreción de potasio es inferior a 30 mmol/l y el TTKG es bajo, la hipocalemia está causada por una pérdida extrarrenal o un desplazamiento transcelular de K+.
Cuando se confirma una pérdida renal excesiva de potasio en un paciente con hipocalemia, el diagnóstico posterior se basa en el pH y la presión arterial. En muchos casos estos simples estudios serán suficientes para el diagnóstico. Un problema importante de creciente interés es el aldosteronismo primario (AP). Algunos estudios clínicos han sugerido que el AP es la causa de más del 10% de la hipertensión arterial (HTA), y es más frecuente en pacientes con HTA resistente a los agentes antihipertensivos.19 La hipopotasemia es uno de los síntomas «clásicos» del AP, pero no es tan frecuente como se consideraba.20 Sin embargo, dado que no existe un patrón de oro en el diagnóstico del AP, merece la pena recordar el eje RAA en pacientes con HTA e hipopotasemia.19
Diagnóstico de la hipomagnesemia
La hipomagnesemia es una anomalía frecuente en los pacientes con GS, aunque no se observa en todos los casos.21 En los pacientes normomagnesémicos con GS, la manifestación clínica y las anomalías electrolíticas son más leves.22
Al igual que los iones de potasio, los de magnesio son filtrados libremente por los glomérulos. El 10% del Mg2+ filtrado es absorbido por el túbulo proximal, el 50-70% en la rama ascendente del asa de Henle. La reabsorción distal depende de los canales epiteliales de Mg2+ TRPM6. La magnesiuria depende de la ingesta oral y debe calcularse el FeMg para establecer el desgaste renal de Mg2+ (FeMg=(magnesio en orina×creatinina sérica)/×100). Un FeMg inferior al 2% sugiere una ingesta deficiente, pérdidas gastrointestinales o desplazamiento de Mg hacia las células. El FeMg superior al 4% sugiere un desgaste renal de Mg2+. Cuando se demuestra el desgaste renal de Mg, debe calcularse la relación Ca/Cr en orina. La relación Ca/Cr en orina de 0,3 es típica en la hipermagesuria del síndrome de Bartter, así como la hipomagnesemia recesiva aislada con normocalciuria, la hipomagnesemia familiar con hipercalciuria y nefrocalcinosis, la hipocalcemia autosómica dominante con hipercalciuria, el tratamiento con diuréticos de asa y la nefropatía causada por algunas nefrotoxinas.23
La homeostasis del magnesio y del potasio están relacionadas entre sí, y la depleción de potasio no puede corregirse hasta la corrección de la hipomagnesemia.23 En promedio, los pacientes deben recibir hasta 500mEq de potasio, 4-5mg/kg/día de 5-10mg de cloruro de magnesio. La amilorida (5-10mg/día) y el espironolactón (200-300mg) son útiles.3
Los síntomas clínicos y los resultados de laboratorio son esenciales para el diagnóstico. En los casos con fenotipo típico, algunos autores recomiendan realizar una prueba de tiazidas para confirmar el diagnóstico.24 En el caso presentado, la investigación genética reveló dos nuevas mutaciones del gen SLC12A3. Probablemente son causadas por la falta de mutaciones funcionales de la PTNC.
Conclusiones
El síndrome de GS es una de las causas raras de hipopotasemia, y parece un reto para los médicos. Mostramos en este trabajo que si uno recuerda sobre un enfoque muy simple de la hipocalemia y es consciente de la acción de los diuréticos y su similitud con las tubulopatías heredadas, el diagnóstico podría ser bastante sencillo. La GS debe diferenciarse de otras tubulopatías (tanto heredadas como adquiridas) y de otras causas de hipopotasemia (por ejemplo, la enfermedad de Conn). Los antecedentes familiares pueden revelar pacientes asintomáticos con GS. Un tratamiento adecuado protege a los pacientes de complicaciones potencialmente peligrosas.