
Una breve introducción a ChIP-Seq

Las interacciones proteína-ADN se utilizan ampliamente para dilucidar los mecanismos subyacentes a la fisiología celular. El desarrollo de la tecnología de ensayo de inmunoprecipitación de la cromatina (ChIP) permitió el estudio de dichos mecanismos. Tras su desarrollo, surgió una alternativa de secuenciación profunda (ChiP-Seq), que ofrece ventajas en términos de especificidad y sensibilidad.
Un experimento de ChIP-Seq comienza con una reticulación de células enteras con formaldehído, seguida de sonicación y aislamiento del ADN. Después se realiza la inmunoprecipitación del complejo ADN-proteína, que consiste en la unión de anticuerpos a proteínas específicas. Los inmunocomplejos formados se precipitan y se purifican. Por último, se secuencia el ADN, generando datos de alta resolución de los sitios enriquecidos. Este enfoque, junto con una línea bien establecida de ChIP-seq, permite a los investigadores capturar los factores de transcripción del ADN, los sitios de modificación de las histonas, las alteraciones epigenéticas y las firmas de la red de regulación de los genes.
Relevancia clínica y aplicaciones
Los desequilibrios epigenéticos en las enfermedades y los estados de salud pueden implicar la modificación de las histonas y la alteración de los factores de transcripción. En este sentido, los estudios ChIP-Seq se han utilizado para dilucidar los mecanismos moleculares patológicos que subyacen al cáncer y otras enfermedades. El análisis ChIP-seq también contribuye a la comprensión del papel de los factores de transcripción durante las enfermedades. De hecho, algunos transcritos parecen estar alterados durante las manifestaciones del fenotipo clínico.
Resumen del pipeline de ChIP-Seq
El pipeline de análisis de ChIP-Seq es el componente principal de los proyectos de interacción ADN-proteína y consta de varios pasos, incluyendo el procesamiento de datos brutos, el análisis de control de calidad, la alineación con el genoma de referencia, la comprobación de la calidad de las lecturas alineadas, la llamada de picos, la anotación y la visualización. Sin embargo, tener un diseño experimental bien pensado es crucial para obtener resultados de alta calidad en un experimento de ChIP-seq. Antes de comenzar su análisis, es esencial tener en cuenta parámetros como las réplicas de muestras, los grupos de control, los kits de secuenciación y las plataformas de secuenciación.
Control de calidad
Todos los informes de Basepair proporcionan puntuaciones de calidad para ayudar a descubrir posibles problemas de secuenciación o contaminación en sus datos de entrada.
El paso de control de calidad (QC) tiene como objetivo evaluar la calidad de los datos de alto rendimiento generados por la secuenciación. Este paso es similar a los realizados en los análisis de DNA-seq y RNA-seq. Aquí, las principales métricas evaluadas incluyen la calidad de la secuencia y de las bases, el contenido de GC, la presencia de adaptadores de secuenciación y las secuencias sobrerrepresentadas. Uno de los programas más utilizados para este tipo de análisis es FastQC. Además, si se identifican secuencias de baja calidad, pueden eliminarse posteriormente durante el paso de recorte. Aunque es un paso opcional, el recorte mejora la calidad de los datos al retener sólo las lecturas de alta calidad.
Alineación
Tras la medición del QC, las lecturas de ChIP-Seq se alinean con un genoma de referencia. El mapeo de lecturas permite a los investigadores identificar el origen de una secuencia de lectura en el genoma. Las herramientas de software de alineación más utilizadas son Bowtie y BWA, ambas utilizadas en las líneas de ChIP-seq de Basepair. Ambas herramientas comparan secuencias poco divergentes con un genoma de referencia.
El flujo de recuento de lecturas ayuda a proporcionar una visión general de las lecturas utilizables al final de los procesos de recorte, alineación y deduplicación. Piense en la figura como una línea de ensamblaje de análisis de datos: entrada de datos en bruto, obtención de una salida de lecturas utilizables.
Comprobación de la calidad de las lecturas alineadas
El siguiente paso consiste en la inferencia de control de calidad del conjunto de datos alineados. Durante el proceso de mapeo, los duplicados de lecturas introducidos por la amplificación de la PCR y la secuenciación causan sesgos durante la llamada de picos y el análisis de enriquecimiento. Basepair utiliza la herramienta Picard para eliminar los duplicados. Una vez eliminados los duplicados, debe evaluar la fracción no redundante (NRF) de las lecturas alineadas. La NRF mide las lecturas únicas que se corresponden con el genoma de referencia. Los experimentos ChIP-seq ideales deberían tener menos de tres lecturas por posición.
Llamada de picos
El paso de llamada de picos detecta las regiones de interacción proteína-ADN enriquecidas en el genoma. El pipeline ChIP-seq de Basepair utiliza MACS2 para realizar este análisis. En MACS2, la llamada de picos se realiza en base a tres pasos principales: estimación de fragmentos, seguida de la identificación de los parámetros de ruido local y luego la identificación de picos. Como resultado de este paso, los usuarios obtienen una tabla final con información sobre los picos, como la puntuación de enriquecimiento, el valor -log10p, el valor -log10q y la posición del inicio del pico. En este paso se recomienda encarecidamente el uso de muestras de control para compararlas con el conjunto de datos objetivo investigado. Tenga en cuenta que los buenos grupos de control aportan resultados más fiables.
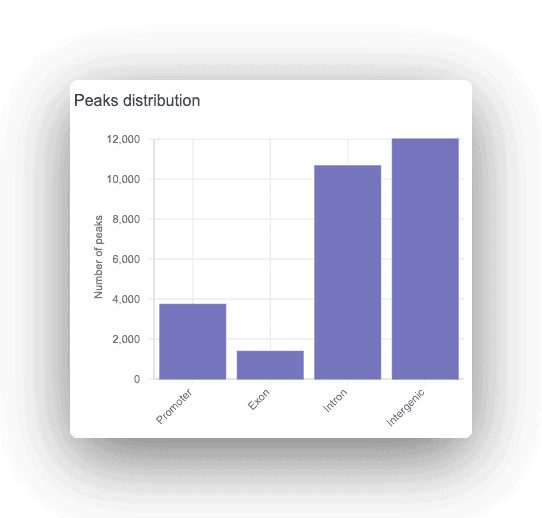
Cada pico se anota como promotor, intrónico o intergénico, y se muestra el gen correspondiente. Para cualquier pico encontrado, se realiza un análisis de motivos para encontrar sitios de unión a factores de transcripción sobrerrepresentados.
Resumen de resultados
Un pipeline de ChIP-seq puede proporcionar no sólo información sobre el estado de la cromatina, sino también sobre la unión a factores de transcripción en un determinado gen o contexto de loci. La aparición de modificaciones de las histonas y de los factores de transcripción en las regiones reguladoras del ADN puede constituir una firma epigenética específica del estado. Así, las perturbaciones epigenéticas pueden asociarse con fenotipos clínicos. Por ejemplo, la heterogeneidad de los estados de la cromatina puede conducir a la resistencia al tratamiento en el cáncer de mama. Estas células tienden a perder los marcadores de modificaciones histónicas represivas y aumentan aún más la expresión de genes que se sabe que promueven la resistencia al tratamiento del cáncer.
Análisis de picos, motivos y vías en la línea de análisis ChIP-Seq
La identificación del enriquecimiento de factores de transcripción por motivos se utiliza para dilucidar si los factores de transcripción están cooperando o compitiendo en una región determinada. La identificación de picos en regiones con motivos de ADN puede mejorar la interpretación de los resultados experimentales. Juntos, tanto el análisis de picos como el de motivos dan una idea de lo que puede estar ocurriendo dentro de una célula. La integración de los enriquecimientos de picos y motivos da como resultado un paisaje epigenómico con posibles consecuencias biológicas. Además, el análisis de vías se utiliza para identificar las proteínas de una vía. Se formulan investigaciones y conclusiones basadas en la presencia de una proteína.
Visualización de datos
Los datos resultantes de un pipeline ChIP-seq pueden ser visualizados usando un navegador de genoma. Los informes de Basepair incluyen un navegador del genoma IGV2 incrustado que le permite interactuar con sus datos. Los datos pueden ser visualizados alternativamente usando heatmaps, que son infografías de intensidad representativa basadas en la densidad de datos que muestran la presencia o ausencia de marcas específicas. Otros gráficos utilizados aquí son el gráfico de enriquecimiento, el upSet y el gráfico de cobertura, que calcula y muestra la cobertura de las regiones de pico en el genoma.
El navegador del genoma es una gran herramienta para visualizar sus datos genómicos en bruto. Está incorporado en cada informe de análisis ChIP-seq en Basepair.
1. Grosselin, K., A. Durand, et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet, v.51, n.6, Jun, p.1060-1066. 2019.
2. Northrup, D. L. e K. Zhao. Aplicación de ChIP-Seq y técnicas relacionadas al estudio de la función inmune. Immunity, v.34, n.6, Jun 24, p.830-42. 2011.
3. Park, S. J., J. H. Kim, et al. A ChIP-Seq Data Analysis Pipeline Based on Bioconductor Packages. Genomics Inform, v.15, n.1, Mar, p.11-18. 2017.
4. Pepke, S., B. Wold, et al. Computation for ChIP-seq and RNA-seq studies. Nat Methods, v.6, n.11 Suppl, Nov, p.S22-32. 2009.
5. Satoh, J., N. Kawana, et al. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul Syst Bio, v.7, p.139-52. 2013.