
Bevezetés
A hipokalémia gyakori klinikai probléma az endokrinológusok és nefrológusok gyakorlatában. A hypokalaemiának számos nyilvánvaló oka van, mint például a hasmenés, a hányás vagy a diuretikumok visszaélése. Más okok, mint például a tubulopátiák ritkán figyelhetők meg, és diagnózisuk nagyobb kihívást jelent. Számos öröklött és szerzett tubulopátia okoz hypokalaemiát, amelyek néha súlyosak és életveszélyesek.1
A hypokalaemia viszonylag gyakori, de figyelmen kívül hagyott oka a Gitelman-szindróma (GS).2 Ez egy recesszív sóvesztő tubulopátia, amelyet az SLC12A3 génmutáció okoz. Az SLC12A3 gén a tiazid-érzékeny transzportert, az NCCT-t (nátrium-klorid ko-transzporter) kódolja. Az NCCT a disztális konvolutált tubuláris sejtekben (DCC) található, amelyek az elektrolitok tubuláris felszívódásának 7-10%-áért felelősek.3
A GS-ben talált legsúlyosabb laboratóriumi eltérések a vese K+ és Mg2+ pazarlása által okozott hypokalaemia és hypomagnesaemia. További jellemző elváltozások a metabolikus alkalózis, a hipokalciuria és a hyperreninémiás hyperaldosteronizmus.4 Gyakran megfigyelhető enyhe vagy közepesen súlyos hipofoszfatémia.5 Súlyos hipofoszfatémiáról is beszámoltak, súlyos hyponatremia mellett.6,7,7
A GS első tünetei olyan gyermekeknél vagy fiatal felnőtteknél jelentkeznek, akik normális növekedéssel rendelkeznek, és a kórtörténetben sóimádó viselkedést mutatnak (a gyermekek mohón fogyasztják a savanyúságot vagy sós sót, sózott uborkát, narancsot és citromot, a gyermekek nyalogatják a sót a burgonyachipszekből stb.8 A klinikai megjelenés betegenként változó. Egyesek tünetmentesek, másoknál azonban életveszélyes szövődmények alakulnak ki. A férfiaknál súlyosabb a fenotípus, mint a nőknél.8 A leggyakoribb tünetek az izomgörcsök és -gyengeség, székrekedés, nokturia, poliuria, szomjúság, polydipsia, szívritmuszavarok, paresztéziák és fokozott sóéhség. Az artériás hipotenzió gyakori és sok esetben a legmarkánsabb tünet, azonban az idősödő GS-populációban hipertónia is előfordulhat.8 A biokémiai eltérések és a tünetek közötti összefüggés nem szoros.9 A GS nem befolyásolja a gyermekek hangulatát és társas kapcsolatait.9,10 Egyébként a tünetek a felnőtteknél gyakoribbak, és negatív hatással lehetnek az életminőségükre. A GS-betegek 45 százaléka mérsékelt vagy nagy problémának tartja a tüneteit.11 A szélsőséges kimerültség, izomgyengeség, paresztéziák, súlyos fáradtság és hipotenzió a mindennapi tevékenységek enyhe vagy súlyos csökkenésével jár.9
A GS becsült prevalenciája 1:40 0008 , a heterozigóta betegek előfordulása pedig legalább 1% az európai populációban. Az SLC12A3 több mint 180 különböző mutációját írták le eddig.12
Egy esetleírás
Egy 26 éves férfi került kórházba syncope, generalizált és izomgyengeség és izomgörcsök előfordulása miatt. A beteg anamnéziséből kiderült, hogy 3,16 mmol/l káliumszint mellett szinkópé epizódot szenvedett. A járóbeteg-szakrendelésen végzett további követés során ismétlődő hypokalaemia előfordulását (a legalacsonyabb érték 2,6 mmol/l) észlelték. A vérnyomás normális volt 110/80, a szívfrekvencia 72/perc; a fizikális vizsgálatban nem volt változás. Neurológiai lelet sem, testsúlya 74 kg, magassága 178 cm. A kórházi felvételkor a laborvizsgálat enyhe hypokalaemiát (K+ 3,0mmol/l), hypomagnesaemiát (Mg2+ 1,36mg/dl), hypocalciuriát (
mg/24h) és metabolikus alkalózist (HCO3- 29,7mmol/l, BE 5,3mmol/l) mutatott. A vesefunkció jó volt eGFR>60ml/perc értékkel. A képalkotó vizsgálatok nem voltak feltűnőek, ahogy az EKG is.
A további vizsgálatok megerősítették, hogy a hypokalaemiát a vese káliumpazarlása okozta. A 24 órás káliumpazarlás, a transtubuláris K+ gradiens (TTKG), a frakcionált K+-kiválasztás (FeK) és a random K/kreatinin arány (K/Cr) a hyperkaliuriára jellemző volt, és az 1. táblázatban látható.13-15 A napi magnesiuria 64mg/24h volt, a frakcionált magnézium (FeMg) kiválasztás 15%.
A hipokalémia diagnosztikus értékei.13-15
| A nem megfelelő kaliurézis diagnosztikus értékei | Páciens | |
|---|---|---|
| Húgyúti K+ | >20mmol/24h Ha a szérum K+3.5mmol/l |
57mmol/l (szérum K+=3.0mmol/l) |
| FeK=×100% | >6,5% Ha szérum K+3.5mmol/l |
12% (szérum K+=3.0mmol/l) |
| TTKG= | TTKG>2 Ha szérum K+3.5mmol/l |
7,3 (szérum K+=3,0mmol/l) |
| uK/Cr – random vizelet K+ és kreatinin aránya | uK/Cr>15mmol/g Ha szérum K+3.5mmol/l |
67mmol/g (szérum K+=3.0mmol/l) |
Bár a vérnyomás normális volt, a Conn-kór kizárására hormonális vizsgálatokat és a hasi CT-t végeztek, és nem mutattak ki semmilyen rendellenességet a mellékvesékben. A szekunder hyperaldoszteronizmus az aldoszteron 289pg/ml (normál tartomány 20-180) és a renin 205mIU/ml (normál tartomány 2,8-39,9) szintjével a GS-re jellemző volt.
A beteget a klinikai fenotípus alapján GS-nek diagnosztizálták. A diagnózis felállítása után kálium- és magnéziumpótló kezelést vezettek be. 20 mmol kálium-kloridot és 18 mmol magnézium-pirrolidon-karboxilátot kapott, klinikai javulással. A családi anamnézis a beteg 35 éves nővérénél enyhe tünetmentes hipokalémiát mutatott ki. A vizsgálatok azonban sem hyperkaliuriát, sem hypomagnesaemiát nem mutattak ki. A GS-t ebben az esetben nem diagnosztizálták.
Genetikai elemzés
A Nozu16 által javasolt genetikai elemzés, algoritmus szerint az SLC12A3 gént hipokalémiás metabolikus alkalózisban szenvedő, teljes korban született, normál súlyú, nefrokalcinózis nélküli, hypocalcuriás és hypomagnesaemiás betegeknél kell vizsgálni.
A leírt esetben genetikai vizsgálatot végeztek, és két heterozigóta mutációt: c.35_36insA és c.1095+5G>A mutációt találtak az SLC12A3 gén NM_000339.2 transzkriptumában (1. ábra). Mindkét mutáció még nem szerepel a HGMD(r) adatbázis elérhető változatában . Az első mutációt a beteg édesanyjában is megtalálták, a másodikat pedig az apában. A betegünkben azonosított két c.35_36insA mutáció közül csak az egyiket találták meg a nővérében.
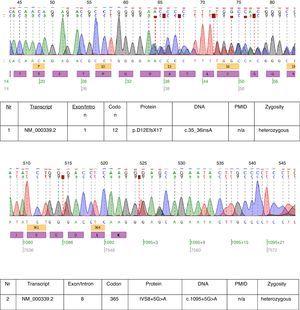
Az érintett beteg DNS-szekvenálásának kromatogramjai.
A betegnél talált mutációk szinte bizonyítják a GS klinikai diagnózisát. A frameshift mutáció patogenetikai jelentősége nyilvánvaló. A 8-as intron splice site mutációjának patogenetikai relevanciájában nem lehetünk biztosak. A 8-as intron splice donorjánál a konszenzusos splice szekvencia G (100%), T (100%), A (62%), A (68%), G (82%), T (63%) nem pontosan egyezik a konszenzusos GTgAGc-vel. Az ebben a betegben talált mutáció tovább tér el a szekvenciában a konszenzustól (GtgAac). A mutációs kóstolóval végzett in silico elemzés is ezt a mutációt betegséget okozónak tartja.17
Diszkusszió
A tubulopátiák ritka betegségek. A RenalTube adatbázis18 szerint az európai populációban a leggyakoribb primer tubulopátiák a distalis renális tubuláris acidózis, a Bartter-szindróma, a familiáris hypomagnesaemia hypercalciuriával és a GS.18 A GS prevalenciája körülbelül 25 eset per 1 millió.3 Mivel a GS az egyik leggyakoribb, valószínűleg sok nefrológus és endokrinológus fog szembesülni GS-esetekkel pályája során. Betegünknél a GS tipikus klinikai megjelenése volt, és jól reagált a terápiára. A GS problémája, hogy a figyelmen kívül hagyott hipokalémia szívmegállás vagy légzőizombénulás miatt halált okozhat. A súlyos neuromuszkuláris tünet, mint a hipokalémiás bénulás a betegek akár 6%-ánál is előfordul (ázsiai betegeknél gyakoribb).3
Az öröklött tubulopátiáknak van egyfajta “tükörképe”, a diuretikumok által okozott szerzett tubulopátiák. Minden diuretikum (pontosabban: natriuretikumok, acquaretikumok vagy glükuretikumok) az öröklött tubulopátiákhoz hasonló rendellenességeket okoz (2. táblázat). Szinte minden tubulopátiában poliuria lép fel, és a káliumszint-változások a leggyakoribb problémák közé tartoznak. A GS-ben szenvedő betegeknél az SLC12A3 gén mutációit találjuk. Ez a gén a tiazid-érzékeny transzportert kódolja. Ezért a GS hasonlít a tiazidokkal való krónikus kezelésre.
Diuretikumok és tubulopátiák.
| Egyetlen gén öröklődő hibája | Káliumkoncentráció | Tünetek hasonló használata: | Gén | Géntermék | Géntermék helye | |
|---|---|---|---|---|---|---|
| Guibaud- | ||||||
| Guibaud-Vainsel-szindróma (RTA 3) | ↓ | Acetazolamid | CA2 | CA II | Proximalis tubulus | . |
| Renális glikozúria | – | SGLT2-gátlóksa (Canagliflozin, Tofogliflozin, Dapagliflozin) | SLC5A2 | Nátrium/glükóz kotranszporter 2 (SGLT2) | Proximális tubulus | |
| Bartter-szindróma | ↓ | Furoszemid | SLC12A1b | Na-K-2Cl kotranszporter (NKCC2) | Henle-hurok | |
| Gitelman-szindróma | ↓ | Tiazid | SLC12A3 | Tiazid-érzékeny Na-Cl kotranszporter | Distalis tubulus | |
| Diabetes insipidus | – | Vaptan | AVPR2, AQP2 | V2 receptor | Collecting ductus | |
| Pseudohypoaldosteronizm, AD | ← | Spironolakton | NR3C2 | Spironolakton-érzékeny mineralokortikoid receptor | Collecting ductus | |
| Pseudohypoaldosteronizm, AR | Amilorid | SCNN1A, SCNN1B, SCNN1G | Amilorid-érzékeny epithelialis nátriumcsatorna | Collecting ductus | ||
AR – autoszomális recesszív; AD – autoszomális domináns.
Az SGLT2-gátlók (glükuretikumok) a 2-es típusú cukorbetegség kezelésének új gyógyszerei; a kezelés során ozmotikus diurézissel kapcsolatos események, mint a pollakiuria és a poliuria figyelhetők meg.
Bár többféle Bartter-gén létezik, ez az, amelyik a furosemidet utánozza.
A hipokalémia megközelítése
AGS-t familiáris hipokalémiás hipomagnezaemiának is nevezik, mivel a hipokalémia a leggyakoribb jelenség. Hypokalaemiás betegnél és tubulopátia gyanúja esetén meg kell erősíteni a hyperkaliuriát. A renális káliumpazarlás TKKG, FeK, random K/Cr arány kiszámításával vagy 24 órás vizeletgyűjtés után bizonyítható (1. táblázat). A kaliuria értékelését akkor kell elvégezni, ha a beteg nem szed sem diuretikumot, sem káliumpótlást, és ha a káliumszint alacsony. Ha a káliumkiválasztás 30 mmol/l alatt van, és a TTKG alacsony, a hypokalaemiát extrarenalis veszteség vagy transzcelluláris K+-eltolódás okozza.
Ha a hypokalaemiás betegnél a túlzott kálium veseelvesztése igazolódik, a további diagnózis a pH és a vérnyomás alapján történik. Sok esetben ezek az egyszerű vizsgálatok elegendőek a diagnózishoz. Egy egyre nagyobb érdeklődésre számot tartó fontos probléma a primer aldoszteronizmus (PA). Egyes klinikai tanulmányok szerint a PA az artériás hipertónia (AH) több mint 10%-ának oka, és gyakoribb az antihipertenzív szerekkel szemben rezisztens AH-s betegeknél.19 A hypokalaemia a PA egyik “klasszikus” tünete, de nem olyan gyakori, mint gondolták.20 Mivel azonban nincs arany standard az AP diagnózisában, érdemes megemlékezni az RAA tengelyről AH-s és hypokalaemiás betegeknél.19
A hypomagnesaemia diagnózisa
A hypomagnesaemia gyakori eltérés a GS-es betegeknél, bár nem minden esetben figyelhető meg.21 A normomagnéziás GS-es betegeknél a klinikai manifesztáció és az elektrolit-rendellenességek enyhébbek.22
A káliumionokhoz hasonlóan a magnéziumionokat is szabadon szűrik a glomerulusok. A szűrt Mg2+ 10%-a a proximális tubulusban, 50-70%-a a Henle-hurok felszálló végében szívódik fel. A distalis reabszorpció a hám Mg2+ TRPM6 csatornáitól függ. A magnesiuria az orális beviteltől függ, és a renális Mg2+-pazarlás megállapításához FeMg-et kell számítani (FeMg=(vizelet magnézium×szérum kreatinin)/×100). A 2%-nál kisebb FeMg alacsony bevitelre, GI-veszteségre vagy a Mg sejtekbe történő átvitelére utal. A 4% feletti FeMg a vese Mg2+ pazarlására utal. Ha bizonyított a renális Mg-pazarlás, random Ca/Cr arányt kell kiszámítani. A vizelet Ca/Cr aránya 0,3 jellemző a Bartter-szindróma hypermagesuriájában, így az izolált recesszív hypomagnesaemia normocalciuriával, a familiáris hypomagnesaemia hypercalciuriával és nephrocalcinosisszal, az autoszomális domináns hypocalcaemia hypercalciuriával, loop diuretikum kezelés és egyes nephrotoxinok által okozott nephropathia esetén.23
A magnézium és a kálium homeosztázis összefügg egymással, és a káliumdepletió nem korrigálható a hypomagnesaemia korrekciójáig.23 A betegeknek átlagosan legfeljebb 500mEq káliumot, 4-5mg/kg/nap 5-10mg magnézium-kloridot kell kapniuk. Az amilorid (5-10mg/nap) és a spironolakton (200-300mg) hasznos.3
A diagnózishoz elengedhetetlenek a klinikai tünetek és a laboratóriumi eredmények. Tipikus fenotípusú esetekben egyes szerzők tiazid teszt elvégzését javasolják a diagnózis megerősítésére.24 A bemutatott esetben a genetikai vizsgálat az SLC12A3 gén két új mutációját mutatta ki. Ezeket valószínűleg az NCCT funkcionális mutációinak hiánya okozza.
Következtetések
A GS-szindróma a hypokalaemia egyik ritka oka, és kihívást jelent az orvosok számára. Ebben a közleményben megmutatjuk, hogy ha valaki emlékszik a hypokalaemia nagyon egyszerű megközelítéséről, és tisztában van a diuretikumok hatásával és az öröklött tubulopátiákhoz való hasonlóságával, a diagnózis meglehetősen egyszerű lehet. A GS-t meg kell különböztetni más (öröklött és szerzett) tubulopátiáktól, valamint a hypokalaemia egyéb okaitól (pl. Conn-kór). A családi anamnézis feltárhat tünetmentes GS-betegeket. A megfelelő kezelés megvédi a betegeket a potenciálisan veszélyes szövődményektől.