
A ChIP-Seq rövid bevezetése

A fehérje-DNS kölcsönhatásokat széles körben használják a sejtfiziológia hátterében álló mechanizmusok feltárására. A kromatin immunprecipitációs (ChIP) assay technológia kifejlesztése lehetővé tette az ilyen mechanizmusok vizsgálatát. A továbbfejlesztést követően kialakult egy mélyszekvenálási alternatíva (ChiP-Seq), amely a specificitás és az érzékenység tekintetében előnyöket kínál.
A ChIP-Seq kísérlet egész sejtek formaldehiddel történő keresztkötésével kezdődik, amelyet szonikáció és DNS-izolálás követ. Ezt követően a DNS-fehérje komplex immunprecipitációja következik, amely specifikus fehérjékhez kötődő antitestekből áll. A képződött immunkomplexeket kicsapjuk és tisztítjuk. Végül a DNS-t szekvenálják, nagy felbontású adatokat generálva a feldúsult helyekről. Ez a megközelítés a jól bevált ChIP-seq csővezetékkel együtt lehetővé teszi a kutatók számára a DNS-transzkripciós faktorok, hiszton-módosítási helyek, epigenetikai változások és génszabályozó hálózati szignatúrák rögzítését.
Klinikai jelentőség és alkalmazások
A betegségek és egészségi állapotok epigenetikai egyensúlytalanságai hiszton-módosítással és megváltozott transzkripciós faktorokkal járhatnak. Itt a ChIP-Seq-vizsgálatokat a rák és más betegségek hátterében álló patológiai molekuláris mechanizmusok feltárására használták. A ChIP-seq-elemzés hozzájárul a transzkripciós faktorok betegségek során betöltött szerepének megértéséhez is. Úgy tűnik ugyanis, hogy egyes transzkriptek megváltoznak a klinikai fenotípus megnyilvánulásai során.
A ChIP-Seq pipeline áttekintése
A ChIP-Seq elemzési pipeline a DNS-fehérje kölcsönhatási projektek fő összetevője, és több lépésből áll, beleértve a nyers adatok feldolgozását, a minőségellenőrzési elemzést, a referencia genomhoz való igazítást, az igazított olvasatok minőségellenőrzését, a csúcsok hívását, az annotációt és a vizualizációt. A ChIP-seq-kísérletekben a kiváló minőségű eredmények eléréséhez azonban elengedhetetlen az átgondolt kísérleti tervezés. Az elemzés megkezdése előtt feltétlenül figyelembe kell venni az olyan paramétereket, mint a mintaismétlések, kontrollcsoportok, szekvenáló készletek és szekvenáló platformok.
Minőségellenőrzés
Minden Basepair jelentés minőségi pontszámot ad, amely segít feltárni a lehetséges szekvenálási problémákat vagy szennyeződéseket a bemeneti adatokban.
A minőségellenőrzési (QC) lépés célja a szekvenálásból származó nagy áteresztőképességű adatok minőségének értékelése. Ez a lépés hasonló a DNS-seq és RNS-seq elemzések során végzett lépésekhez. Itt a fő értékelt metrikák közé tartozik a szekvencia- és bázisminőség, a GC-tartalom, a szekvenálási adaptorok jelenléte és a felülreprezentált szekvenciák. Az ilyen típusú elemzésekhez az egyik leggyakrabban használt program a FastQC. Ezen túlmenően, ha alacsony minőségű szekvenciákat azonosítanak, ezek később eltávolíthatók a trimmelési lépés során. Bár ez egy opcionális lépés, a trimmelés javítja az adatok minőségét azáltal, hogy csak a jó minőségű leolvasásokat tartja meg.
Alignment
A QC-mérés után a ChIP-Seq leolvasásokat egy referencia genomhoz igazítják. A leolvasás leképezése lehetővé teszi a kutatók számára, hogy azonosítsák a leolvasott szekvencia eredetét a genomban. A népszerű illesztési szoftvereszközök közé tartozik a Bowtie és a BWA, amelyeket a Basepair ChIP-seq pipeline-okban is használnak. Mindkét eszköz alacsony divergens szekvenciákat térképez fel egy referencia genommal szemben.
A leolvasásszám-áramlás segít a használható leolvasások átfogó áttekintését biztosítani a trimmelési, igazítási és deduplikációs folyamatok végén. Gondoljon az ábrára úgy, mint egy adatelemzési futószalagra: bemeneti nyers adatok, kimenetként használható olvasatokat kap.
Az igazított olvasatok minőségellenőrzése
A következő lépés az igazított adathalmaz QC következtetéséből áll. A leképezési folyamat során a PCR-amplifikáció és a szekvenálás által bevezetett olvasatduplikátumok torzításokat okoznak a csúcsmegjelölés és a dúsítási elemzés során. A Basepair a Picard eszközt használja a duplikátumok eltávolítására. A duplikátumok eltávolítása után ki kell értékelni az illesztett olvasatok nem redundáns frakcióját (NRF). Az NRF a referencia genomhoz tartozó egyedi leolvasások számát méri. Az ideális ChIP-seq-kísérleteknek pozíciónként háromnál kevesebb olvasatot kell tartalmazniuk.
Peak Calling
A peak calling lépés a genomban feldúsult fehérje-DNS kölcsönhatási régiókat detektálja. A Basepair ChIP-seq csővezetéke a MACS2-t használja ennek az elemzésnek az elvégzéséhez. A MACS2-ben a csúcshívás három fő lépésen alapul: a fragmentumbecslés, majd a helyi zajparaméterek azonosítása, végül a csúcsok azonosítása. E lépés kimeneteleként a felhasználók egy végső táblázatot kapnak a csúcsinformációkkal, például a gazdagodási pontszámmal, a -log10pértékkel, a -log10qértékkel és a csúcs kezdetéhez viszonyított pozícióval. Ebben a lépésben erősen ajánlott a kontrollminták használata a vizsgált céladatkészlettel való összehasonlításhoz. Ne feledje, hogy a jó kontrollcsoportok megbízhatóbb eredményeket hoznak.
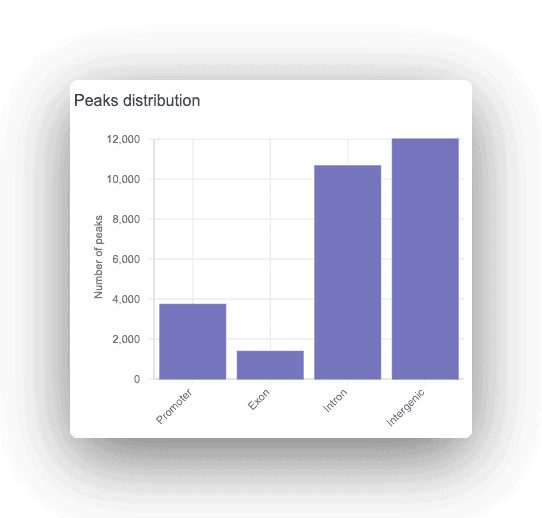
Minden csúcsot promóterként, intronikus vagy intergenikusként annotáltunk, a megfelelő gén megjelenítésével. Minden talált csúcs esetében motívumelemzést végeznek, hogy megtalálják a felülreprezentált transzkripciós faktor-kötőhelyeket.
Eredmények áttekintése
A ChIP-seq pipeline nem csak a kromatin állapotáról adhat információt, hanem a transzkripciós faktorok kötődéséről is egy meghatározott gén vagy lokusz kontextusában. A hisztonmódosulások és a transzkripciós faktorok DNS-szabályozó régiókban való előfordulása állapot-specifikus epigenetikai szignatúrát alkothat. Így az epigenetikai perturbációk összefüggésbe hozhatók a klinikai fenotípusokkal. A kromatinállapotok heterogenitása például mellrák esetén kezelési rezisztenciához vezethet. Ezek a sejtek hajlamosak elveszíteni a represszív hisztonmódosulások markereit, és tovább növelik a rákkezeléssel szembeni rezisztenciát elősegítő ismert gének expresszióját.
Peak, Motif és Pathway Analysis in ChIP-Seq Analysis Pipeline
A motívumos transzkripciós faktorok feldúsulásának azonosítása annak tisztázására szolgál, hogy a transzkripciós faktorok együttműködnek vagy versengenek-e egy adott régióban. A DNS-motívumos régiókban lévő csúcsok azonosítása javíthatja a kísérleti eredmények értelmezését. A csúcs- és motívumelemzések együttesen betekintést nyújtanak abba, hogy mi történhet egy sejtben. A csúcs- és motívumgazdagodások integrációja egy epigenomikai tájképet eredményez, amelynek lehetséges biológiai következményei vannak. Továbbá az útvonalelemzés egy útvonalban lévő fehérjék azonosítására szolgál. A vizsgálatok és következtetések egy-egy fehérje jelenléte alapján kerülnek megfogalmazásra.
Adatvizualizáció
A ChIP-seq csővezetékből származó adatok genomböngésző segítségével vizualizálhatók. A Basepair jelentések tartalmaznak egy beágyazott IGV2 genom böngészőt, amely lehetővé teszi az adatokkal való interakciót. Az adatokat alternatív módon heatmaps segítségével is lehet vizualizálni, amelyek az adatsűrűségen alapuló reprezentatív intenzitás-infografikák, amelyek az egyes jelölések jelenlétét vagy hiányát mutatják. Az itt használt egyéb grafikonok közé tartozik az enrichment plot, az upSet és a coverage plot, amely egyszerre számítja ki és jeleníti meg a csúcsrégiók lefedettségét a genomban.
A genom böngésző nagyszerű eszköz a nyers genomi adatok vizualizálásához. Be van építve a Basepair minden ChIP-seq-elemzési jelentésébe.
1. Grosselin, K., A. Durand, et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet, v.51, n.6, Jun, p.1060-1066. 2019.
2. Northrup, D. L. e K. Zhao. A ChIP-Seq és a kapcsolódó technikák alkalmazása az immunfunkció vizsgálatára. Immunity, v.34, n.6, Jun 24, p.830-42. 2011.
3. Park, S. J., J. H. Kim, et al. A ChIP-Seq Data Analysis Pipeline Based on Bioconductor Packages. Genomics Inform, v.15, n.1, Mar, p.11-18. 2017.
4. Pepke, S., B. Wold, et al. Computation for ChIP-seq and RNA-seq studies. Nat Methods, v.6, n.11 Suppl, Nov, p.S22-32. 2009.
5. Satoh, J., N. Kawana, et al. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul Syst Bio, v.7, p.139-52. 2013.