
Wprowadzenie
Hypokaliemia jest częstym problemem klinicznym w praktyce endokrynologów i nefrologów. Istnieje wiele oczywistych przyczyn hipokaliemii, takich jak biegunka, wymioty czy nadużywanie diuretyków. Inne przyczyny, takie jak tubulopatie, są rzadko obserwowane, a ich diagnostyka stanowi większe wyzwanie. Istnieje wiele dziedzicznych i nabytych tubulopatii powodujących hipokaliemię, czasami ciężką i zagrażającą życiu.1
Stosunkowo częstą, ale pomijaną przyczyną hipokaliemii jest zespół Gitelmana (GS).2 Jest to recesywna tubulopatia z utratą soli spowodowana mutacją genu SLC12A3. Gen SLC12A3 koduje wrażliwy na tiazydy transporter NCCT (sodium chloride co-transporter). NCCT znajduje się w komórkach kanalika dystalnego (distal convoluted tubular cells – DCC), które odpowiadają za 7-10% kanalikowego wchłaniania elektrolitów.3
Najcięższymi nieprawidłowościami laboratoryjnymi występującymi w GS są hipokaliemia i hipomagnezemia spowodowane nerkowym marnotrawstwem K+ i Mg2+. Inne typowe zmiany to zasadowica metaboliczna, hipokalciuria i hiperaldosteronizm hipreninemiczny.4 Często obserwuje się łagodną do umiarkowanej hipofosfatemię.5 Opisywano również ciężką hipofosfatemię z ciężką hiponatremią.6,7
Pierwsze objawy GS występują u dzieci lub młodych dorosłych z prawidłowym wzrostem i wywiadem dotyczącym zachowań związanych z łaknieniem soli (dzieci chętnie spożywające ogórki kiszone lub solankę, solone ogórki, pomarańcze i cytryny, dzieci zlizujące sól z chipsów ziemniaczanych itp. U niektórych przebiega bezobjawowo, ale u innych rozwijają się zagrażające życiu powikłania. Mężczyźni wykazują cięższy fenotyp niż kobiety.8 Najczęstszymi objawami są skurcze i osłabienie mięśni, zaparcia, nokturia, wielomocz, pragnienie, polidypsja, zaburzenia rytmu serca, parestezje i zwiększony apetyt na sól. Niedociśnienie tętnicze jest częstym i w wielu przypadkach najbardziej widocznym objawem, jednak w starzejącej się populacji chorych na GS może wystąpić nadciśnienie tętnicze.8 Korelacja między nieprawidłowościami biochemicznymi a objawami nie jest silna.9 GS nie zaburza nastroju i relacji społecznych u dzieci.9,10 W przeciwnym razie objawy występują częściej u dorosłych i mogą mieć negatywny wpływ na jakość ich życia. Czterdzieści pięć procent pacjentów z GS uważa swoje objawy za umiarkowany lub duży problem.11 Skrajne wyczerpanie, osłabienie mięśni, parestezje, ciężkie zmęczenie i niedociśnienie tętnicze wiążą się z łagodnym lub ciężkim ograniczeniem codziennej aktywności.9
Oszacowana częstość występowania GS wynosi 1:40 0008 , a częstość występowania heterozygotycznego wynosi co najmniej 1% w populacji europejskiej. Dotychczas opisano ponad 180 różnych mutacji w SLC12A3.12
Raport przypadku
26-letni mężczyzna został przyjęty do szpitala z powodu występowania omdleń, uogólnionego i mięśniowego osłabienia oraz skurczów mięśni. W wywiadzie pacjent podawał epizod omdleń z poziomem potasu 3,16 mmol/l. W dalszej obserwacji w warunkach ambulatoryjnych obserwowano nawracające przypadki hipokaliemii (najniższa wartość 2,6 mmol/l). Ciśnienie tętnicze krwi było prawidłowe 110/80, częstość akcji serca 72/min; w badaniu przedmiotowym nie stwierdzono zmian. Nie stwierdzono również zmian neurologicznych, waga 74 kg, wzrost 178 cm. Przy przyjęciu do szpitala w ocenie laboratoryjnej stwierdzono łagodną hipokaliemię (K+ 3,0mmol/l), hipomagnezemię (Mg2+ 1,36mg/dl), hipokalciurię (
mg/24h) oraz zasadowicę metaboliczną (HCO3- 29,7mmol/l, BE 5,3mmol/l). Czynność nerek była dobra z eGFR>60ml/min. Badania obrazowe były bez zmian, podobnie jak EKG.
Dalsze badania potwierdziły, że hipokaliemia jest spowodowana nerkową utratą potasu. 24-godzinne marnowanie potasu, przezcewkowy gradient K+ (TTKG), frakcyjne wydalanie K+ (FeK) i przypadkowy stosunek K/kreatynina (K/Cr) były typowe dla hiperkaliemii i przedstawiono je w Tabeli 1.13-15 Dobowa magneziuria wynosiła 64mg/24h, z frakcyjnym wydalaniem magnezu (FeMg) 15%.
Wartości diagnostyczne dla hipokaliemii.13.-15
| Wartości diagnostyczne dla niewłaściwej kaliurezy | Pacjent | |
|---|---|---|
| Urinary K+ | >20mmol/24h Jeśli surowica K+3.5mmol/l |
57mmol/l (surowica K+=3.0mmol/l) |
| FeK=×100% | >6,5% Jeśli surowica K+3.5mmol/l |
12% (serum K+=3,0mmol/l) |
| TTKG= | TTKG>2 If serum K+3.5mmol/l |
7,3 (surowica K+=3,0mmol/l) |
| uK/Cr – losowy stosunek K+ do kreatyniny w moczu | uK/Cr>15mmol/g Jeśli surowica K+3.5mmol/l |
67mmol/g (surowica K+=3.0mmol/l) |
Ale ciśnienie krwi było prawidłowe, wykonano badania hormonalne w celu wykluczenia choroby Conna oraz CT jamy brzusznej, które nie wykazały żadnych nieprawidłowości w nadnerczach. Wtórny hiperaldosteronizm z poziomem aldosteronu 289pg/ml (zakres prawidłowy 20-180) i reniny 205mIU/ml (zakres prawidłowy 2,8-39,9) był typowy dla GS.
Pacjentka została zdiagnozowana jako GS na podstawie fenotypu klinicznego. Po postawieniu diagnozy wdrożono leczenie suplementacją potasu i magnezu. Pacjent otrzymywał 20 mmol chlorku potasu i 18 mmol karboksylanu pirolidonu magnezu, uzyskując poprawę kliniczną. W wywiadzie rodzinnym stwierdzono łagodną bezobjawową hipokaliemię u 35-letniej siostry pacjenta. Badania nie wykazały jednak hiperkaliemii ani hipomagnezemii. Nie rozpoznano GS w tym przypadku.
Analiza genetyczna
Zgodnie z analizą genetyczną, algorytmem zaproponowanym przez Nozu16 gen SLC12A3 powinien być badany u pacjentów z hipokaliemiczną zasadowicą metaboliczną, z urodzeniem o czasie, z prawidłową masą ciała bez nefrokalcynozy, z hipokalcemią i hipomagnezemią.
W opisanym przypadku wykonano badania genetyczne i stwierdzono dwie heterozygotyczne mutacje: c.35_36insA i c.1095+5G>A w transkrypcie NM_000339.2 genu SLC12A3 (ryc. 1). Obie mutacje nie znajdują się jeszcze w dostępnej wersji bazy danych HGMD(r) . Pierwszą mutację stwierdzono również u matki pacjentki, a drugą u ojca. Tylko jedna z dwóch mutacji zidentyfikowanych u naszego pacjenta c.35_36insA została znaleziona u jego siostry.
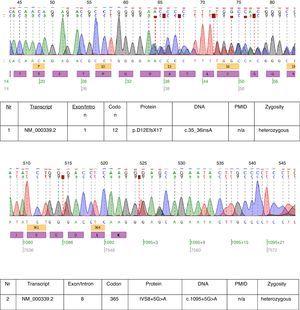
Chromatogramy sekwencjonowania DNA pacjenta dotkniętego chorobą.
Mutacje znalezione u tego pacjenta niemalże potwierdzają kliniczne rozpoznanie GS. Znaczenie patogenetyczne mutacji frameshift jest oczywiste. Nie możemy być pewni co do znaczenia patogenetycznego mutacji miejsca splice w intronie 8. Konsensus sekwencji splotu G (100%), T (100%), A (62%), A (68%), G (82%), T (63%) u donora splotu intronu 8 nie odpowiada dokładnie konsensusowi GTgAGc. Mutacja znaleziona u tego pacjenta jeszcze bardziej odchyla sekwencję od konsensusu (GtgAac). Również analiza in silico przeprowadzona przez mutation taster uważa tę mutację za powodującą chorobę.17
Dyskusja
Tubulopatie są rzadkimi chorobami. Według bazy danych RenalTube18 najczęstszymi pierwotnymi tubulopatiami w populacji europejskiej są: dystalna nerkowa kwasica cewkowa, zespół Barttera, rodzinna hipomagnezemia z hiperkalciurią i GS.18 Częstość występowania GS wynosi około 25 przypadków na 1 milion.3 Ponieważ GS jest jedną z najczęstszych, prawdopodobnie wielu nefrologów i endokrynologów będzie miało do czynienia z przypadkami GS w trakcie swojej kariery zawodowej. Nasz pacjent miał typowy obraz kliniczny GS i dobrze zareagował na leczenie. Problemem w GS jest to, że przeoczona hipokaliemia może spowodować zgon z powodu zatrzymania krążenia lub porażenia mięśni oddechowych. Ciężki objaw nerwowo-mięśniowy, jakim jest porażenie hipokaliemiczne, występuje nawet u 6% pacjentów (częściej u pacjentów z Azji).3
Dziedziczone tubulopatie mają swoiste „lustrzane odbicia” – nabyte tubulopatie wywołane przez diuretyki. Każdy z diuretyków (a dokładnie: natriuretyki, acquaretyki i glukurtyki) powoduje zaburzenia podobne do tych, które występują w tubulopatiach dziedzicznych (tab. 2). Niemal w każdej tubulopatii występuje wielomocz, a zmiany stężenia potasu należą do najczęstszych problemów. U pacjentów z GS stwierdza się mutacje genu SLC12A3. Gen ten koduje transporter wrażliwy na tiazydy. Dlatego GS przypomina przewlekłe leczenie tiazydami.
Diuretyki i tubulopatie.
| Wrodzony defekt pojedynczego genu | Stężenie potasu | Objawy podobne do stosowania: | Gen | Produkt genu | Lokalizacja produktu genu |
|---|---|---|---|---|---|
| Guibaud-.Zespół Vainsela (RTA 3) | ↓ | Acetazolamid | CA2 | CA II | Cewka proksymalna |
| Glikozuria nerkowa | – | Inhibitory SGLT2a (Canagliflozin, Tofogliflozin, Dapagliflozin) | SLC5A2 | Kotransporter sodu/glukozy 2 (SGLT2) | Kanalik proksymalny |
| Zespół Barttera | ↓ | Furosemid | SLC12A1b | Na-.K-2Cl cotransporter (NKCC2) | Pętla Henlego |
| Zespół Gitelmana | ↓ | Tiazyd | SLC12A3 | Tiazyd.sensitive Na-Cl cotransporter | Distal tubule |
| Diabetes insipidus | – | Vaptan | AVPR2, AQP2 | Receptor V2 | Kanał zbiorczy |
| Pseudohypoaldosteronizm, AD | ← | Spironolakton | NR3C2 | Wrażliwy na ironolakton receptor mineralokortykoidowy | Kolektor |
| Pseudohypoaldosteronizm, AR | Amiloryd | SCNN1A, SCNN1B, SCNN1G | Wrażliwy na amiloryd nabłonkowy kanał sodowy | Kanał zbiorczy | |
AR – autosomalny recesywny; AD – autosomalny dominujący.
Inhibitory SGLT2 (glukurtyki) są nowymi lekami w leczeniu cukrzycy typu 2; podczas leczenia obserwuje się zdarzenia związane z diurezą osmotyczną, takie jak pollakiuria i poliuria.
Choć istnieją różne geny Barttera, to ten właśnie gen naśladuje furosemid.
Podejście do hipokaliemii
GS jest również znana jako rodzinna hipokaliemia hipomagnezowa, ponieważ hipokaliemia jest najczęstszym zjawiskiem. U pacjenta z hipokaliemią i podejrzeniem tubulopatii należy potwierdzić hiperkaliemię. Nerkową stratę potasu można wykazać, obliczając TKKG, FeK, losowy stosunek K/Cr lub po 24-godzinnej zbiórce moczu (tab. 1). Ocenę kaliurii należy przeprowadzać, gdy pacjent nie przyjmuje diuretyków ani suplementacji potasu oraz gdy stężenie potasu jest niskie. Gdy wydalanie potasu wynosi poniżej 30 mmol/l, a TTKG jest niskie, hipokaliemia jest spowodowana utratą pozanerkową lub przezkomórkowym przesunięciem K+.
Gdy potwierdzona jest nadmierna utrata nerkowa potasu u pacjenta z hipokaliemią, dalsza diagnostyka opiera się na badaniu pH i ciśnienia tętniczego. W wielu przypadkach te proste badania wystarczą do postawienia rozpoznania. Ważnym problemem budzącym coraz większe zainteresowanie jest pierwotny aldosteronizm (PA). Niektóre badania kliniczne sugerują, że PA jest przyczyną ponad 10% nadciśnienia tętniczego (arterial hypertension, AH) i występuje częściej u chorych z AH opornym na leki przeciwnadciśnieniowe.19 Hipokaliemia jest jednym z „klasycznych” objawów PA, ale nie jest tak częsta, jak uważano.20 Ponieważ jednak nie ma złotego standardu w rozpoznawaniu AP, warto pamiętać o osi RAA u chorych z AH i hipokaliemią.19
Diagnostyka hipomagnezemii
Hypomagnezemia jest częstą nieprawidłowością u pacjentów z GS, choć nie obserwuje się jej w każdym przypadku.21 U normomagnezemicznych pacjentów z GS objawy kliniczne i zaburzenia elektrolitowe są łagodniejsze.22
Podobnie jak jony potasu, magnezowe są swobodnie filtrowane przez kłębuszki nerkowe. 10% przefiltrowanego Mg2+ jest wchłaniane w kanaliku proksymalnym, 50-70% we wstępującym odcinku pętli Henlego. Dystalna reabsorpcja zależy od nabłonkowych kanałów Mg2+ TRPM6. Magnezuria zależy od ilości przyjmowanego doustnie magnezu, a FeMg powinien być obliczony w celu ustalenia nerkowego niedoboru Mg2+ (FeMg=(magnez w moczu×kreatynina w surowicy)/×100). FeMg poniżej 2% sugeruje słabe spożycie, straty w żołądku lub przesunięcie Mg do komórek. FeMg powyżej 4% sugeruje nerkową utratę Mg2+. W przypadku wykazania nerkowej utraty Mg należy obliczyć losowo stosunek Ca/Cr. Stosunek Ca/Cr w moczu 0,3 jest typowy dla hipermagesurii w zespole Barttera, podobnie jak izolowana recesywna hipomagnezemia z normokalciurią, rodzinna hipomagnezemia z hiperkalciurią i nefrokalcynozą, autosomalna dominująca hipokalcemia z hiperkalciurią, leczenie diuretykami pętlowymi i nefropatia wywołana przez niektóre nefrotoksyny.23
Homeostaza magnezu i potasu są ze sobą powiązane, a deplecja potasu nie może być skorygowana do czasu skorygowania hipomagnezemii.23 Przeciętnie pacjenci powinni otrzymywać do 500mEq potasu, 4-5mg/kg/dobę 5-10mg chlorku magnezu. Pomocne są amiloryd (5-10mg/dobę) i spironolakton (200-300mg).3
Objawy kliniczne i wyniki badań laboratoryjnych są kluczowe dla rozpoznania. W przypadkach o typowym fenotypie niektórzy autorzy zalecają wykonanie testu tiazydowego w celu potwierdzenia rozpoznania.24 W prezentowanym przypadku badania genetyczne wykazały dwie nowe mutacje genu SLC12A3. Prawdopodobnie są one spowodowane brakiem funkcjonalnych mutacji NCCT.
Wnioski
ZespółGS jest jedną z rzadkich przyczyn hipokaliemii i stanowi wyzwanie dla lekarzy. W niniejszej pracy pokazujemy, że jeśli pamięta się o bardzo prostym podejściu do hipokaliemii i jest się świadomym działania diuretyków oraz ich podobieństwa do dziedzicznych tubulopatii, rozpoznanie może być dość proste. GS powinna być różnicowana z innymi tubulopatiami (zarówno dziedzicznymi, jak i nabytymi) oraz innymi przyczynami hipokaliemii (np. choroba Conna). Wywiad rodzinny może ujawnić bezobjawowych pacjentów z GS. Odpowiednie leczenie chroni pacjentów przed potencjalnie groźnymi powikłaniami.