
Krótkie wprowadzenie do ChIP-Seq

Interakcje białko-DNA są szeroko wykorzystywane do wyjaśnienia mechanizmów leżących u podstaw fizjologii komórki. Rozwój technologii testów immunoprecypitacji chromatyny (ChIP) umożliwił badanie tych mechanizmów. Po dalszym rozwoju, powstała alternatywa głębokiego sekwencjonowania (ChiP-Seq), która oferuje korzyści w zakresie specyficzności i czułości.
Eksperyment ChIP-Seq rozpoczyna się od usieciowania całej komórki formaldehydem, po czym następuje sonikacja i izolacja DNA. Następnie przeprowadzana jest immunoprecypitacja kompleksu DNA-białko, która polega na wiązaniu się przeciwciał ze specyficznymi białkami. Powstałe immunokompleksy są wytrącane i oczyszczane. Na koniec DNA jest sekwencjonowane, generując dane o wysokiej rozdzielczości wzbogaconych miejsc. To podejście, wraz z dobrze rozwiniętym potokiem ChIP-seq, pozwala badaczom na wychwycenie czynników transkrypcyjnych DNA, miejsc modyfikacji histonów, zmian epigenetycznych i sygnatur sieci regulacyjnych genów.
Relewantność kliniczna i zastosowania
Zaburzenia równowagi epigenetycznej w chorobach i stanach zdrowia mogą obejmować modyfikacje histonów i zmienione czynniki transkrypcyjne. W tym kontekście, badania ChIP-Seq zostały wykorzystane do wyjaśnienia patologicznych mechanizmów molekularnych leżących u podstaw raka i innych chorób. Analiza ChIP-seq przyczynia się również do zrozumienia roli czynników transkrypcyjnych w procesach chorobowych. W rzeczywistości, niektóre transkrypty wydają się być zmienione podczas manifestacji fenotypu klinicznego.
Overview of the ChIP-Seq pipeline
Potok analizy ChIP-Seq jest głównym składnikiem projektów interakcji DNA-białko i składa się z kilku etapów, w tym przetwarzania surowych danych, analizy kontroli jakości, wyrównania do genomu referencyjnego, kontroli jakości wyrównanych odczytów, wywoływania pików, anotacji i wizualizacji. Jednakże, posiadanie przemyślanego projektu eksperymentu jest kluczowe dla uzyskania wysokiej jakości wyników w eksperymencie ChIP-seq. Przed rozpoczęciem analizy należy rozważyć parametry takie jak repliki próbek, grupy kontrolne, zestawy do sekwencjonowania i platformy sekwencjonowania.
Kontrola jakości
Wszystkie raporty Basepair dostarczają wyników jakości, aby pomóc w wykryciu potencjalnych problemów z sekwencjonowaniem lub zanieczyszczeń w danych wejściowych.
Krok kontroli jakości (QC) ma na celu ocenę jakości danych o wysokiej przepustowości wygenerowanych z sekwencjonowania. Ten krok jest podobny do tych wykonywanych w analizach DNA-seq i RNA-seq. Głównymi ocenianymi metrykami są tutaj jakość sekwencji i baz, zawartość GC, obecność adaptorów sekwencjonowania oraz nadreprezentowane sekwencje. Jednym z najczęściej używanych programów do tego typu analiz jest FastQC. Ponadto, w przypadku zidentyfikowania sekwencji o niskiej jakości, mogą być one później usunięte podczas etapu przycinania. Chociaż jest to krok opcjonalny, przycinanie poprawia jakość danych poprzez zachowanie tylko wysokiej jakości odczytów.
Przyrównanie
Po pomiarze QC, odczyty ChIP-Seq są wyrównywane do genomu referencyjnego. Mapowanie odczytów pozwala badaczom zidentyfikować pochodzenie sekwencji odczytu w genomie. Do popularnych narzędzi programowych służących do wyrównywania należą Bowtie i BWA, które są używane w potokach ChIP-seq firmy Basepair. Oba narzędzia mapują nisko rozbieżne sekwencje względem genomu referencyjnego.
Przepływ liczby odczytów pomaga uzyskać duży obraz użytecznych odczytów na końcu procesów przycinania, wyrównywania i deduplikacji. Potraktuj tę figurę jako linię montażową analizy danych: wprowadź surowe dane, uzyskaj wynik użytecznych odczytów.
Kontrola jakości wyrównanych odczytów
Następny krok obejmuje wnioskowanie QC wyrównanego zbioru danych. Podczas procesu mapowania, duplikaty odczytów wprowadzone przez amplifikację PCR i sekwencjonowanie powodują błędy podczas wywoływania pików i analizy wzbogacania. Basepair wykorzystuje narzędzie Picard do usuwania duplikatów. Po usunięciu duplikatów należy ocenić frakcję nieredukowalną (NRF) wyrównanych odczytów. NRF mierzy unikalne odczyty mapujące do genomu referencyjnego. Idealne eksperymenty ChIP-seq powinny mieć mniej niż trzy odczyty na pozycję.
Wywoływanie szczytów
Krok wywoływania szczytów wykrywa wzbogacone regiony interakcji białko-DNA w genomie. Potoczek ChIP-seq firmy Basepair używa MACS2 do wykonania tej analizy. W MACS2, wywoływanie pików jest wykonywane w oparciu o trzy główne kroki: estymację fragmentów, następnie identyfikację lokalnych parametrów szumu i identyfikację pików. Jako wynik tego etapu użytkownicy otrzymują końcową tabelę z informacjami o piku, takimi jak wynik wzbogacenia, wartość -log10p, wartość -log10q i pozycja do początku piku. Użycie próbek kontrolnych jest wysoce zalecane w tym kroku dla porównania z badanym zbiorem danych docelowych. Należy pamiętać, że dobre grupy kontrolne przynoszą bardziej wiarygodne wyniki.
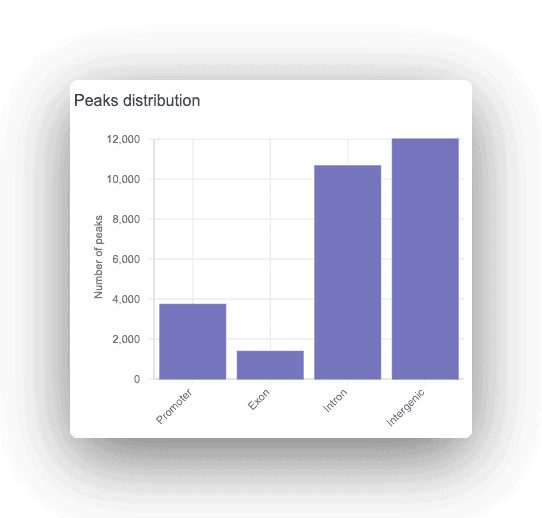
Każdy pik jest opisywany jako promotor, intronowy lub intergeniczny, z wyświetlonym odpowiadającym mu genem. Dla każdego znalezionego piku przeprowadzana jest analiza motywów w celu znalezienia nadreprezentowanych miejsc wiązania czynników transkrypcyjnych.
Overview of Results
Potok ChIP-seq może dostarczyć nie tylko informacji o stanie chromatyny, ale również o wiązaniu czynników transkrypcyjnych w kontekście określonego genu lub loci. Występowanie modyfikacji histonów i czynników transkrypcyjnych w regionach regulacyjnych DNA może stanowić specyficzną dla danego stanu sygnaturę epigenetyczną. W ten sposób perturbacje epigenetyczne mogą być związane z fenotypami klinicznymi. Na przykład, heterogenność stanów chromatyny może prowadzić do oporności na leczenie w raku piersi. Komórki te mają tendencję do utraty represyjnych markerów modyfikacji histonów i dalszego zwiększania ekspresji genów znanych z promowania oporności na leczenie nowotworów.
Peak, Motif and Pathway Analysis in ChIP-Seq Analysis Pipeline
Identyfikacja wzbogacenia czynnika transkrypcyjnego w motywy jest wykorzystywana do wyjaśnienia, czy czynniki transkrypcyjne współpracują czy konkurują w danym regionie. Identyfikacja pików w regionach motywów DNA może poprawić interpretację wyników eksperymentalnych. Łącznie, zarówno analizy pików jak i motywów dają wgląd w to, co może zachodzić w komórce. Integracja wzbogacenia pików i motywów daje w efekcie krajobraz epigenomowy z możliwymi konsekwencjami biologicznymi. Ponadto, analiza szlaków jest używana do identyfikacji białek w szlaku. Badania i wnioski są formułowane w oparciu o obecność białek.
Wizualizacja danych
Dane wynikowe z potoku ChIP-seq mogą być wizualizowane przy użyciu przeglądarki genomu. Raporty Basepair zawierają wbudowaną przeglądarkę genomu IGV2, która pozwala na interakcję z danymi. Dane mogą być również wizualizowane przy użyciu map cieplnych, które są infografikami o reprezentatywnej intensywności opartymi na gęstości danych, które pokazują obecność lub brak określonych znaczników. Inne grafiki stosowane tutaj to wykres wzbogacenia, wykres upSet i wykres pokrycia, który zarówno oblicza jak i wyświetla pokrycie regionów szczytowych w genomie.
Przeglądarka genomu jest doskonałym narzędziem do wizualizacji surowych danych genomowych. Jest ono wbudowane w każdy raport analizy ChIP-seq w Basepair.
1. Grosselin, K., A. Durand, et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet, v.51, n.6, Jun, s.1060-1066. 2019.
2. Northrup, D. L. e K. Zhao. Application of ChIP-Seq and related techniques to the study of immune function. Immunity, v.34, n.6, Jun 24, s.830-42. 2011.
3. Park, S. J., J. H. Kim, et al. A ChIP-Seq Data Analysis Pipeline Based on Bioconductor Packages. Genomics Inform, v.15, n.1, Mar, s.11-18. 2017.
4. Pepke, S., B. Wold, et al. Computation for ChIP-seq and RNA-seq studies. Nat Methods, v.6, n.11 Suppl, Nov, p.S22-32. 2009.
5. Satoh, J., N. Kawana, et al. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul Syst Bio, v.7, s.139-52. 2013.