
Introducere
Hipopotasemia este o problemă clinică frecventă în practica endocrinologilor și nefrologilor. Există multe cauze evidente de hipokaliemie, cum ar fi diareea, vărsăturile sau abuzul de diuretice. Alte cauze, cum ar fi tubulopatiile, sunt rareori observate, iar diagnosticul lor este mai dificil. Există multe tubulopatii moștenite și dobândite care cauzează hipopotasemie, uneori severă și amenințătoare de viață.1
O cauză relativ frecventă, dar trecută cu vederea, de hipopotasemie este sindromul Gitelman (GS).2 Este o tubulopatie recesivă cu pierdere de sare cauzată de mutația genei SLC12A3. Gena SLC12A3 codifică transportorul sensibil la tiazide NCCT (co-transportor de clorură de sodiu). NCCT este localizat în celulele tubulare convolutive distale (DCC), care sunt responsabile pentru 7-10% din absorbția tubulară a electroliților.3
Cele mai severe anomalii de laborator întâlnite în GS sunt hipokaliemia și hipomagneziemia cauzate de pierderea renală de K+ și Mg2+. Alte modificări tipice sunt alcaloza metabolică, hipocalciuria și hiperaldosteronismul hiperreninemic.4 Se observă frecvent hipofosfatemie ușoară până la moderată.5 S-a raportat, de asemenea, hipofosfatemie severă cu hiponatremie severă.6,7
Primele simptome ale GS apar la copiii sau adulții tineri cu creștere normală și antecedente de comportamente de apetit pentru sare (copii dornici să consume murături sau saramură, castraveți, portocale și lămâi sărate, copii care ling sarea de pe cartofii prăjiți, etc.).8 Prezentarea clinică variază în funcție de pacienți. Unii sunt asimptomatici, dar alții dezvoltă complicații care le amenință viața. Bărbații manifestă un fenotip mai sever decât femeile.8 Cele mai frecvente simptome sunt crampe și slăbiciune musculară, constipație, nocturie, poliurie, sete, polidipsie, aritmii cardiace, parestezii și apetit crescut pentru sare. Hipotensiunea arterială este frecventă și, în multe cazuri, cel mai proeminent simptom, cu toate acestea, în populația GS îmbătrânită poate apărea hipertensiune arterială.8 Corelația dintre anomaliile biochimice și simptome nu este puternică.9 GS nu interferează cu starea de spirit și relațiile sociale ale copiilor.9,10 În caz contrar, simptomele sunt mai frecvente la adulți și pot avea un impact negativ asupra calității vieții acestora. Patruzeci și cinci la sută dintre pacienții cu GS consideră că simptomele lor reprezintă o problemă moderată până la mare.11 Epuizarea extremă, slăbiciunea musculară, parestezii, oboseala severă și hipotensiunea sunt asociate cu reducerea ușoară până la severă a activităților zilnice.9
Prevalența estimată a GS este de 1:40.0008 , iar prevalența heterozigoților este de cel puțin 1% în populația europeană. Până în prezent au fost descrise peste 180 de mutații diferite în SLC12A3.12
Raport de caz
Un bărbat de 26 de ani a fost internat în spital din cauza incidenței sincopei, slăbiciune generalizată și musculară și crampe musculare. Anamneza pacientului a relevat un episod de sincopă cu un nivel al potasiului de 3,16mmol/l. La urmărirea ulterioară în evaluarea ambulatorie, s-a observat incidența recurentă a hipopotasemiei (cea mai mică valoare 2,6mmol/l). Tensiunea arterială a fost normală 110/80, frecvența cardiacă a fost de 72 pe minut; nu au existat modificări la examenul fizic. Nici constatări neurologice, greutate 74 kg, înălțime 178 cm. La internarea în spital evaluarea de laborator a arătat hipokaliemie ușoară (K+ 3,0mmol/l), hipomagneziemie (Mg2+ 1,36mg/dl), hipocalciurie (
mg/24h) și alcaloză metabolică (HCO3- 29,7mmol/l, BE 5,3mmol/l). Funcția renală a fost bună, cu eGFR>60ml/min. Studiile imagistice au fost neremarcabile, la fel și ECG-ul.
Investigațiile suplimentare au confirmat că hipokaliemia a fost cauzată de irosirea renală a potasiului. Depășirea de potasiu în 24 de ore, gradientul transtubular de K+ (TTKG), excreția fracționată de K+ (FeK) și raportul K/creatinină aleatorie (K/Cr) au fost tipice pentru hiperkaliurie și sunt prezentate în tabelul 1.13-15 Magneziuria zilnică a fost de 64mg/24h, cu o excreție fracționată de magneziu (FeMg) de 15%.
Valorile diagnostice pentru hipokaliemie.13-15
| Valorile de diagnostic pentru o kaliureză inadecvată | Pacient | |
|---|---|---|
| K+ urinar | >20mmol/24h Dacă K+3 seric.5mmol/l |
57mmol/l (K+ seric=3.0mmol/l) |
| FeK=×100% | >6,5% Dacă serul K+3.5mmol/l |
12% (K+ seric=3,0mmol/l) |
| TTKG= | TTKG>2 Dacă K+ seric=3,0mmol/l) |
|
| TTKG= | TTKG>2 Dacă K+3.5mmol/l |
7,3 (K+ seric=3,0mmol/l) |
| uK/Cr – raport aleatoriu K+ urină la creatinină | uK/Cr>15mmol/g Dacă K+3 seric.5mmol/l |
67mmol/g (K+ seric=3.0mmol/l) |
Deși tensiunea arterială a fost normală, au fost efectuate teste hormonale pentru a exclude boala Conn și CT abdominal, care nu au evidențiat anomalii la nivelul glandelor suprarenale. Hiperaldosteronismul secundar cu niveluri de aldosteron 289pg/ml (interval normal 20-180) și renină 205mIU/ml (interval normal 2,8-39,9) au fost tipice pentru GS.
Pacientul a fost diagnosticat ca GS pe baza unui fenotip clinic. După diagnostic, a fost introdus tratament cu suplimente de potasiu și magneziu. A primit 20 mmol de clorură de potasiu și 18 mmol de carboxilat de pirrolidonă de magneziu, cu ameliorare clinică. Anamneza familială a evidențiat hipopotasemie ușoară asimptomatică la sora pacientului, în vârstă de 35 de ani. Dar investigațiile nu au arătat nici hiperkaliurie, nici hipomagneziemie. GS nu a fost diagnosticat în acest caz.
Analiză genetică
Potrivit analizei genetice, algoritmul propus de Nozu16Gena SLC12A3 trebuie testată la pacienții cu alcaloză metabolică hipokaliemică, cu naștere la termen, greutate normală, fără nefrocalcinoză, cu hipocalcemie și hipomagneziemie.
În cazul descris s-a efectuat testarea genetică și s-au constatat două mutații heterozigote: c.35_36insA și c.1095+5G>A au fost găsite în transcriptul NM_000339.2 al genei SLC12A3 (Fig. 1). Ambele mutații nu se regăsesc încă în versiunea disponibilă a bazei de date HGMD(r) . Prima mutație a fost găsită și la mama pacientului, iar cea de-a doua la tată. Doar una dintre cele două mutații identificate la pacientul nostru c.35_36insA a fost găsită la sora sa.
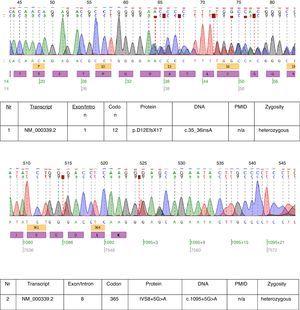
Cromatograme de secvențiere a ADN-ului pacientului afectat.
Mutațiile găsite la acest pacient aproape că dovedesc diagnosticul clinic de GS. Relevanța patogenetică a mutației frameshift este evidentă. Nu putem fi siguri cu privire la relevanța patogenetică a mutației situsului de racordare a intronului 8. Secvența de racordare consensuală G (100%), T (100%), A (62%), A (68%), G (82%), T (63%) la donatorul de racordare al intronului 8 nu se potrivește exact cu consensul GTgAGc. Mutația găsită la acest pacient deviază și mai mult secvența de la consens (GtgAac). De asemenea, analiza in silico prin degustător de mutații consideră această mutație ca fiind cauza bolii.17
Discuție
Tubulopatiile sunt boli rare. Conform bazei de date RenalTube18 , cele mai frecvente tubulopatii primare în populația europeană sunt acidoza tubulară renală distală, sindromul Bartter, hipomagneziemia familială cu hipercalciurie și GS.18 Prevalența GS este de aproximativ 25 de cazuri la 1 milion.3 Deoarece GS este una dintre cele mai frecvente, probabil că mulți nefrologi și endocrinologi se vor confrunta cu cazuri de GS pe parcursul carierei lor. Pacientul nostru a avut o prezentare clinică tipică de GS și a răspuns bine la terapie. Problema cu GS este că hipokaliemia neglijată ar putea cauza decesul din cauza stopului cardiac sau a paraliziei mușchilor respiratori. Simptomul neuromuscular sever, cum ar fi paralizia hipokaliemică, apare la până la 6% dintre pacienți (mai frecvent la pacienții asiatici).3
Tubulopatiile moștenite au un fel de „imagini în oglindă” – tubulopatiile dobândite cauzate de diuretice. Fiecare diuretic (mai exact: natriureticele, achareticele sau glucureticele) determină anomalii asemănătoare cu cele întâlnite în tubulopatiile ereditare (tabelul 2). Aproape în fiecare tubulopatie apare poliurie, iar modificările nivelului de potasiu sunt printre cele mai frecvente probleme. La pacienții cu GS se găsesc mutații ale genei SLC12A3. Această genă codifică transportorul sensibil la tiazide. De aceea GS se aseamănă cu tratamentul cronic cu tiazide.
Diuretice și tubulopatii.
| Defect genetic unic moștenit | Concentrația de potasiu | Simptome similare cu utilizarea de: | Gena | Produs genetic | Localizarea produsului genetic | |
|---|---|---|---|---|---|---|
| Guibaud-Sindromul Vainsel (RTA 3) | ↓ | Acetazolamidă | CA2 | CA II | Tubul proximal | |
| Glicozurie renală | – | Inhibitori SGLT2orsa (Canagliflozin, Tofogliflozin, Dapagliflozin) | SLC5A2 | Cotransportator 2 de sodiu/glucoză (SGLT2) | Tubul proximal | |
| Sindromul Bartter | ↓ | Furosemid | SLC12A1b | Na-K-2Cl cotransportator (NKCC2) | Bucleul Henle | |
| Sindromul Gitelman | ↓ | Tiazidă | SLLC12A3 | Tiazidă-cotransportator Na-Cl sensibil | Tubul distal | |
| Diabetul insipid | – | Vaptan | AVPR2, AQP2 | Receptor V2 | Canal colector | |
| Pseudohipoaldosteronizm, AD | ← | Spironolactona | NR3C2 | Receptor mineralocorticoidic sensibil la spironolactonă | Canalul colector | |
| Pseudohipoaldosteronizm, AR | Amiloridă | SCNN1A, SCNN1B, SCNN1G | Canalul de sodiu epitelial sensibil la amiloridă | Canal de sodiu epitelial | Conductă colectoare | |
AR – autosomal recesiv; AD – autosomal dominant.
Inhibitorii SGLT2 (glucuretice) sunt noile medicamente pentru managementul diabetului zaharat de tip 2; în timpul tratamentului se observă evenimente legate de diureza osmotică, cum ar fi polakiuria și poliuria.
Deși există diferite gene Bartter, aceasta este cea care mimează furosemidul.
Abordarea hipopotasemiei
GS este cunoscută și sub denumirea de hipomagneziemie familială hipopotasemică, deoarece hipopotasemia este fenomenul cel mai frecvent. La un pacient cu hipopotasemie și suspiciune de tubulopatie trebuie să se confirme hiperkaliuria. Depășirea renală a potasiului poate fi dovedită prin calculul TKKG, FeK, raportul K/Cr aleatoriu sau după colectarea urinei de 24 de ore (tabelul 1). Evaluarea kaliuriei trebuie efectuată, atunci când pacientul nu ia nici diuretice, nici suplimente de potasiu și când nivelul de potasiu este scăzut. Atunci când excreția de potasiu este sub 30 mmol/l și TTKG este scăzută, hipokaliemia este cauzată de pierderi extrarenale sau de o deplasare transcelulară de K+.
Când se confirmă pierderea renală excesivă de potasiu la pacientul cu hipokaliemie, diagnosticul ulterior se bazează pe pH și tensiune arterială. În multe cazuri, aceste studii simple vor fi suficiente pentru diagnostic. O problemă importantă de un interes crescând este aldosteronismul primar (AP). Unele studii clinice au sugerat că PA este cauza a peste 10% din hipertensiunea arterială (HTA) și este mai frecventă la pacienții cu HTA rezistentă la agenți antihipertensivi.19 Hipokaliemia este unul dintre simptomele „clasice” ale PA, dar nu este atât de frecventă pe cât s-a considerat.20 Totuși, deoarece nu există un standard de aur în diagnosticul PA, merită să ne amintim despre axa RAA la pacienții cu HTA și hipokaliemie.19
Diagnosticul hipomagneziemiei
Hipomagneziemia este o anomalie frecventă la pacienții cu GS, deși nu este observată în toate cazurile.21 La pacienții normomagneziemici cu GS, manifestarea clinică și anomaliile electrolitice sunt mai ușoare.22
Similare ionilor de potasiu, cei de magneziu sunt filtrați liber de glomeruli. 10% din Mg2+ filtrat este absorbit de tubul proximal, 50-70% în membrul ascendent al ansei lui Henle. Reabsorbția distală depinde de canalele epiteliale Mg2+ TRPM6. Magnesiuria depinde de aportul oral și trebuie calculat FeMg pentru a stabili deficitul renal de Mg2+ (FeMg=(magneziu urinar×creatinină serică)/×100). FeMg mai mică de 2% sugerează un aport deficitar, pierderi GI sau deplasarea Mg în celule. FeMg mai mare de 4% sugerează o pierdere renală de Mg2+. În cazul în care se dovedește o pierdere renală de Mg, trebuie calculat raportul Ca/Cr aleatoriu. Raportul Ca/Cr urinar 0,3 este tipic în hipermagesuria din sindromul Bartter, la fel ca și hipomagnezia recesivă izolată cu normocalciurie, hipomagnezia familială cu hipercalciurie și nefrocalcinoză, hipocalcemia autosomal dominantă cu hipercalciurie, tratamentul cu diuretice de ansă și nefropatia cauzată de unele nefrotoxine.23
Homeostazia magneziului și a potasiului sunt legate una de cealaltă, iar depleția de potasiu nu poate fi corectată până la corectarea hipomagneziemiei.23 În medie, pacienții trebuie să primească până la 500mEq de potasiu, 4-5mg/kg/zi de 5-10mg de clorură de magneziu. Amilorida (5-10mg/zi) și spironolactonul (200-300mg) sunt utile.3
Simptomele clinice și rezultatele de laborator sunt esențiale pentru diagnostic. În cazurile cu fenotip tipic, unii autori recomandă efectuarea unui test tiazidic pentru a confirma diagnosticul.24 În cazul prezentat, investigația genetică a evidențiat două mutații noi ale genei SLC12A3. Acestea sunt probabil cauzate de lipsa mutațiilor funcționale ale NCCT.
Concluzii
Sindromul SG este una dintre cauzele rare de hipokaliemie și pare o provocare pentru medici. Noi arătăm în această lucrare că, dacă se ține minte despre o abordare foarte simplă a hipopotasemiei și se cunoaște acțiunea diureticelor și asemănarea lor cu tubulopatiile ereditare, diagnosticul ar putea fi destul de simplu. GS ar trebui să fie diferențiat de alte tubulopatii (atât moștenite, cât și dobândite), precum și de alte cauze de hipopotasemie (de exemplu, boala Conn). Anamneza familială poate dezvălui pacienți asimptomatici cu GS. Un tratament adecvat protejează pacienții de complicații potențial periculoase.
.