
O scurtă introducere în ChIP-Seq

Interacțiunile proteină-ADN sunt utilizate pe scară largă pentru a elucida mecanismele care stau la baza fiziologiei celulare. Dezvoltarea tehnologiei de testare a imunoprecipitării cromatinei (ChIP) a permis studierea unor astfel de mecanisme. În urma dezvoltării ulterioare, a apărut o alternativă de secvențiere profundă (ChiP-Seq), care oferă avantaje în ceea ce privește specificitatea și sensibilitatea.
Un experiment ChIP-Seq începe cu o reticulare a celulelor întregi cu formaldehidă, urmată de sonicare și de izolarea ADN-ului. După aceea, se realizează imunoprecipitarea complexului ADN-proteină, care constă în legarea anticorpilor la proteine specifice. Imuno-complecșii formați sunt precipitați și purificați. În cele din urmă, ADN-ul este secvențiat, generând date de înaltă rezoluție ale situsurilor îmbogățite. Această abordare, împreună cu un pipeline ChIP-seq bine stabilit, permite cercetătorilor să captureze factorii de transcripție ADN, situsurile de modificare a histonelor, alterările epigenetice și semnăturile rețelelor de reglementare a genelor.
Clinical Relevance and Applications
Dezechilibrele epigenetice în cadrul bolilor și stărilor de sănătate pot implica modificarea histonelor și alterarea factorilor de transcripție. În acest sens, studiile ChIP-Seq au fost utilizate pentru a elucida mecanismele moleculare patologice care stau la baza cancerului și a altor boli. Analiza ChIP-seq contribuie, de asemenea, la înțelegerea rolului factorilor de transcripție în timpul bolilor. De fapt, unii transcripți par a fi modificați în timpul manifestărilor fenotipurilor clinice.
Overview of the ChIP-Seq pipeline
The ChIP-Seq analysis pipeline is the main component of DNA-protein interaction projects and consists of several steps, including raw data processing, quality control analysis, alignment to the reference genome, quality check of the aligned reads, peak calling, annotation, and visualization. Cu toate acestea, existența unui proiect experimental bine gândit este crucială pentru a obține rezultate de înaltă calitate în cadrul unui experiment ChIP-seq. Înainte de a vă începe analiza, este esențial să luați în considerare parametri precum replicile de probe, grupurile de control, kiturile de secvențiere și platformele de secvențiere.
Quality Control
Toate rapoartele Basepair oferă scoruri de calitate pentru a ajuta la descoperirea unor potențiale probleme de secvențiere sau contaminare în datele de intrare.
Etapa Quality Control (QC) are ca scop evaluarea calității datelor de mare randament generate de secvențiere. Această etapă este similară cu cele efectuate în cadrul analizelor ADN-seq și ARN-seq. Aici, principalii parametri evaluați includ calitatea secvențelor și a bazelor, conținutul GC, prezența adaptorilor de secvențiere și secvențele suprareprezentate. Unul dintre cele mai frecvent utilizate programe pentru acest tip de analiză este FastQC. În plus, în cazul în care sunt identificate secvențe de calitate scăzută, acestea pot fi eliminate ulterior în timpul etapei de tundere. Deși este o etapă opțională, tunderea îmbunătățește calitatea datelor prin reținerea doar a citirilor de înaltă calitate.
Alignment
După măsurarea QC, citirile ChIP-Seq sunt aliniate la un genom de referință. Cartografierea citirilor permite cercetătorilor să identifice originea unei secvențe citite în genom. Printre instrumentele software de aliniere populare utilizate se numără Bowtie și BWA, ambele fiind utilizate în conductele ChIP-seq ale Basepair. Ambele instrumente cartografiază secvențe cu divergențe reduse față de un genom de referință.
Fluxul de număr de citiri ajută la furnizarea unei imagini de ansamblu a citirilor utilizabile la sfârșitul proceselor de tăiere, aliniere și deduplicare. Gândiți-vă la această figură ca la o linie de asamblare a analizei de date: introduceți datele brute, obțineți o ieșire de citiri utilizabile.
Controlul de calitate al citirilor aliniate
Postul următor constă în inferența QC a setului de date aliniate. În timpul procesului de cartografiere, dublurile de citire introduse de amplificarea PCR și de secvențiere provoacă distorsiuni în timpul apelării vârfurilor și al analizei de îmbogățire. Basepair utilizează instrumentul Picard pentru a elimina dublurile. Odată ce duplicatele au fost eliminate, ar trebui să evaluați fracția neredundantă (Non-Redundant Fraction – NRF) a citirilor aliniate. NRF măsoară citirile unice care corespund genomului de referință. Experimentele ChIP-seq ideale ar trebui să aibă mai puțin de trei citiri pe poziție.
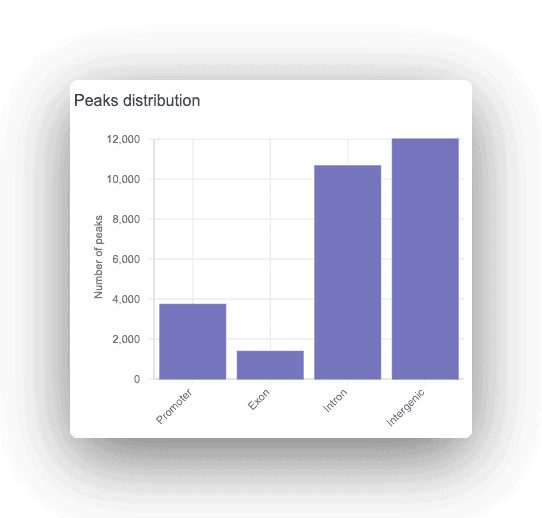
Peak Calling
Etapa de apelare a vârfurilor detectează regiunile de interacțiune proteină-ADN îmbogățite din genom. Conducta ChIP-seq a Basepair utilizează MACS2 pentru a efectua această analiză. În MACS2, peak calling se realizează pe baza a trei etape principale: estimarea fragmentelor, urmată de identificarea parametrilor locali de zgomot și apoi identificarea vârfurilor. Ca rezultat al acestei etape, utilizatorii obțin un tabel final cu informații despre vârfuri, cum ar fi scorul de îmbogățire, valoarea -log10p, valoarea -log10q și poziția față de începutul vârfului. Utilizarea eșantioanelor de control este foarte recomandată în această etapă pentru comparație cu setul de date țintă investigat. Rețineți că grupurile de control bune aduc rezultate mai fiabile.
Care vârf este adnotat ca promotor, intronic sau intergenic, cu gena corespunzătoare afișată. Pentru orice vârfuri găsite, se face o analiză a motivelor pentru a găsi site-urile de legare a factorilor de transcripție suprareprezentate.
Overview of Results
Un pipeline ChIP-seq poate furniza nu numai informații despre starea cromatinei, ci și despre legarea factorilor de transcripție într-un anumit context de gene sau loci. Apariția modificărilor histonice și a factorilor de transcripție în regiunile de reglementare a ADN-ului poate constitui o semnătură epigenetică specifică unei condiții. Astfel, perturbațiile epigenetice pot fi asociate cu fenotipuri clinice. De exemplu, eterogenitatea stărilor de cromatină poate duce la rezistența la tratament în cazul cancerului de sân. Aceste celule au tendința de a pierde markerii de modificări histonice represive și de a crește în continuare expresia genelor cunoscute pentru a promova rezistența la tratamentul cancerului.
Peak, Motif and Pathway Analysis in ChIP-Seq Analysis Pipeline
Identificarea îmbogățirii factorilor de transcripție cu motive este utilizată pentru a elucida dacă factorii de transcripție cooperează sau concurează într-o anumită regiune. Identificarea vârfurilor în regiunile cu motive ADN poate îmbunătăți interpretarea rezultatelor experimentale. Împreună, atât analiza vârfurilor, cât și cea a motivelor oferă informații despre ceea ce se poate întâmpla în interiorul unei celule. Integrarea îmbogățirii vârfurilor și a motivelor are ca rezultat un peisaj epigenomic cu posibile consecințe biologice. În plus, analiza căilor de acces este utilizată pentru a identifica proteinele dintr-o cale. Investigațiile și concluziile sunt formulate pe baza prezenței unei proteine.
Data Visualization
Datele rezultate dintr-o conductă ChIP-seq pot fi vizualizate cu ajutorul unui browser de genom. Rapoartele Basepair includ un browser de genom IGV2 încorporat care vă permite să interacționați cu datele. Datele pot fi vizualizate alternativ folosind heatmaps, care sunt infografice de intensitate reprezentative bazate pe densitatea datelor care arată prezența sau absența unor mărci specifice. Alte grafice utilizate aici includ graficul de îmbogățire, upSet și graficul de acoperire, care calculează și afișează atât acoperirea regiunilor de vârf pe genom.
Browserul genomului este un instrument excelent pentru vizualizarea datelor dvs. genomice brute. Acesta este încorporat în fiecare raport de analiză ChIP-seq de pe Basepair.
1. Grosselin, K., A. Durand, et al. High-throughput single-cell ChIP-seq identifică eterogenitatea stărilor de cromatină în cancerul de sân. Nat Genet, v.51, n.6, iunie, p.1060-1066. 2019.
2. Northrup, D. L. și K. Zhao. Application of ChIP-Seq and related techniques to the study of immune function (Aplicarea ChIP-Seq și a tehnicilor conexe la studiul funcției imune). Immunity, v.34, n.6, Jun 24, p.830-42. 2011.
3. Park, S. J., J. H. Kim, et al. A ChIP-Seq Data Analysis Pipeline Based on Bioconductor Packages. Genomics Inform, v.15, n.1, Mar, p.11-18. 2017.
4. Pepke, S., B. Wold, et al. Computation for ChIP-seq and RNA-seq studies (Calcul pentru studii ChIP-seq și RNA-seq). Nat Methods, v.6, n.11 Suppl, Nov, p.S22-32. 2009.
5. Satoh, J., N. Kawana, et al. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul Syst Bio, v.7, p.139-52. 2013.