
Introduktion
Hypokaliæmi er et almindeligt klinisk problem i endokrinologers og nefrologers praksis. Der er mange indlysende årsager til hypokaliæmi såsom diarré, opkastninger eller misbrug af diuretika. Andre årsager såsom tubulopatier er sjældent observeret, og deres diagnose er mere udfordrende. Der findes mange arvelige og erhvervede tubulopatier, der forårsager hypokaliæmi, som undertiden er alvorlig og livstruende.1
En relativt almindelig, men overset årsag til hypokaliæmi er Gitelman-syndromet (GS).2 Det er en recessiv salttabsgivende tubulopati forårsaget af SLC12A3-genmutationen. SLC12A3-genet koder for den thiazidfølsomme transportør NCCT (natriumklorid co-transporter). NCCT er placeret i de distale konvolutterede tubulære celler (DCC), som er ansvarlige for 7-10 % af den elektrolyt tubulære absorption.3
De mest alvorlige laboratorieafvigelser, der findes i GS, er hypokaliæmi og hypomagnesiæmi forårsaget af renal K+ og Mg2+ svind. Andre typiske ændringer er metabolisk alkalose, hypocalciuri og hyperreninæmisk hyperaldosteronisme.4 Let til moderat hypofosfatæmi observeres hyppigt.5 Der er også rapporteret om alvorlig hypofosfatæmi med alvorlig hyponatriæmi.6,7
De første symptomer på GS forekommer hos børn eller unge voksne med normal vækst og historik med saltkærlig adfærd (børn er ivrige efter at indtage pickles eller saltlage, saltede agurker, appelsiner og citroner, børn slikker salt fra kartoffelchips osv.).8 Den kliniske præsentation varierer fra patient til patient. Nogle er asymptomatiske, men andre udvikler livstruende komplikationer. Mænd manifesterer en mere alvorlig fænotype end kvinder.8 De mest almindelige symptomer er muskelkramper og svaghed, obstipation, nocturia, polyuri, tørst, polydipsi, hjertearytmier, paræstesier og øget saltefterspørgsel. Arteriel hypotension er almindelig og i mange tilfælde det mest fremtrædende symptom, men i den aldrende GS-population kan der forekomme hypertension.8 Korrelationen mellem biokemiske abnormiteter og symptomer er ikke stærk.9 GS forstyrrer ikke børns humør og sociale relationer.9,10 Ellers er symptomer mere almindelige hos voksne og kan have negativ indvirkning på deres livskvalitet. Femogfyrre procent af GS-patienterne betragter deres symptomer som et moderat til stort problem.11 Ekstrem udmattelse, muskelsvaghed, paræstesier, alvorlig træthed og hypotension er forbundet med mild til alvorlig reduktion i daglige aktiviteter.9
Den anslåede prævalens af GS er 1:40.0008 og prævalensen af heterozygote er mindst 1 % i den europæiske befolkning. Mere end 180 forskellige mutationer i SLC12A3 er indtil nu blevet beskrevet.12
Sagsberetning
En 26-årig mand blev indlagt på hospitalet på grund af forekomst af synkope, generaliseret og muskelsvaghed og muskelkramper. Patientens anamnese afslørede en episode af synkope med et kaliumniveau på 3,16 mmol/l. Ved yderligere opfølgning i forbindelse med ambulant vurdering blev der observeret tilbagevendende forekomst af hypokaliæmi (den laveste værdi 2,6 mmol/l). Blodtrykket var normalt 110/80, hjertefrekvensen var 72 pr. minut, og der var ingen ændringer i den fysiske undersøgelse. Ingen neurologiske fund, vægt 74 kg, højde 178 cm. Ved indlæggelse på hospitalet viste laboratorieundersøgelse mild hypokaliæmi (K+ 3,0mmol/l), hypomagnesiæmi (Mg2+ 1,36mg/dl), hypocalciuri (
mg/24h) og metabolisk alkalose (HCO3- 29,7mmol/l, BE 5,3mmol/l). Nyrefunktionen var god med eGFR>60ml/min. Billedundersøgelser var ubemærkede, det samme var EKG’et.
Der blev ved yderligere undersøgelser bekræftet, at hypokaliæmi var forårsaget af renal kaliumsvind. 24-timers kaliumsvind, transtubulær K+ gradient (TTKG), fraktionel K+ udskillelse (FeK) og tilfældigt K/kreatinin-forhold (K/Cr) var typisk for hyperkaliuri og er vist i tabel 1.13-15 Den daglige magnesiuri var 64 mg/24h, med en fraktionel magnesiumudskillelse (FeMg) på 15 %.
Værdierne er diagnostiske for hypokaliæmi.13-15
| Værdier, der er diagnostiske for uhensigtsmæssig kaliurese | Patient | ||||
|---|---|---|---|---|---|
| Urinært K+ | >20mmol/24h Hvis serum K+3.5mmol/l |
57mmol/l (serum K+=3.0mmol/l) |
|||
| FeK=×100% | >6,5% Hvis serum K+3.5mmol/l |
12% (serum K+=3,0mmol/l) |
|||
| TTKG= | TTKG>2 Hvis serum K+3.5mmol/l |
7,3 (serum K+=3,0mmol/l) |
|||
| uK/Cr – tilfældigt K+/kreatinin-forhold i urinen | uK/Cr>15mmol/g Hvis serum K+3.5mmol/l |
67mmol/g (serum K+=3.0mmol/l) |
|||
Og selv om blodtrykket var normalt, blev der foretaget hormonprøver for at udelukke Conn’s sygdom og abdominal CT, som ikke afslørede nogen abnormiteter i binyrerne. Sekundær hyperaldosteronisme med niveauer af aldosteron 289pg/ml (normalområdet 20-180) og renin 205mIU/ml (normalområdet 2,8-39,9) var typisk for GS.
Patienten blev diagnosticeret som GS på baggrund af en klinisk fænotype. Efter diagnosen blev der iværksat behandling med tilskud af kalium og magnesium. Han fik 20 mmol kaliumchlorid og 18 mmol magnesium pyrrolidoncarboxylat med klinisk forbedring. Familieanamnese afslørede mild asymptomatisk hypokaliæmi hos patientens 35-årige søster. Men undersøgelserne viste hverken hyperkaliuri eller hypomagnesiæmi. GS blev ikke diagnosticeret i dette tilfælde.
Genetisk analyse
I henhold til den genetiske analyse, algoritme foreslået af Nozu16SLC12A3-genet bør testes hos patienter med hypokaliæmisk metabolisk alkalose, med fuld term fødsel, normal vægt uden nefrocalcinose, med hypocalcuria og hypomagnesiæmi.
I det beskrevne tilfælde blev der udført genetisk testning, og to heterozygote mutationer: c.35_36insA og c.1095+5G>A blev fundet i transkript NM_000339.2 af SLC12A3-genet (fig. 1). Begge mutationer findes endnu ikke i den tilgængelige HGMD(r)-databaseversion . Den første mutation blev også fundet hos patientens mor og den anden hos faderen. Kun den ene af de to mutationer, der blev identificeret hos vores patient c.35_36insA, blev fundet hos hans søster.
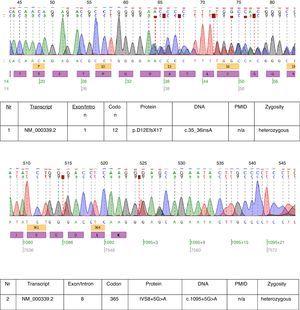
Kromatogrammer af den berørte patients DNA-sekventering.
De mutationer, der blev fundet hos denne patient, beviser næsten den kliniske diagnose af GS. Den patogenetiske relevans af frameshift-mutationen er indlysende. Vi kan ikke være sikre på den patogenetiske relevans af mutationen på splejsestedet i intron 8. Konsensus-splejsesekvensen G (100 %), T (100 %), A (62 %), A (68 %), G (82 %), T (63 %) på splejsedonoren i intron 8 stemmer ikke nøjagtigt overens med konsensus GTgAGc. Den mutation, der er fundet hos denne patient, afviger sekvensen yderligere fra konsensus (GtgAac). Også in silico-analysen ved hjælp af mutationstesteren betragter denne mutation som sygdomsfremkaldende.17
Diskussion
Tubulopatier er sjældne sygdomme. Ifølge RenalTube-databasen18 er de mest almindelige primære tubulopatier i den europæiske befolkning distal renal tubulær acidose, Bartters syndrom, familiær hypomagnesiæmi med hypercalciuri og GS.18 Prævalensen af GS er ca. 25 tilfælde pr. 1 million.3 Da GS er en af de mest almindelige, vil mange nefrologer og endokrinologer sandsynligvis blive konfronteret med tilfælde af GS i løbet af deres karriere. Vores patient havde en typisk klinisk præsentation af GS og reagerede godt på behandlingen. Problemet med GS er, at overset hypokaliæmi kan medføre døden som følge af hjertestop eller lammelse af åndedrætsmusklerne. Det alvorlige neuromuskulære symptom som hypokalæmisk lammelse forekommer hos op til 6 % af patienterne (hyppigere hos asiatiske patienter).3
De arvelige tubulopatier har en slags “spejlbilleder”-erhvervede tubulopatier forårsaget af diuretika. Alle diuretika (for at være præcis: natriuretika, acquaretika eller glucuretika) forårsager abnormiteter, der ligner dem, der findes i arvelige tubulopatier (tabel 2). Næsten i alle tubulopatier forekommer polyuri, og ændringer i kaliumniveauet er blandt de mest almindelige problemer. Hos patienter med GS findes mutationer i SLC12A3-genet. Dette gen koder for den thiazidfølsomme transportør. Derfor ligner GS kronisk behandling med thiazider.
Diuretika og tubulopatier.
| Arvelig enkeltgendefekt | Kaliumkoncentration | Symptomer svarende til brug af: | Gen | Genprodukt | Genproduktets placering | ||
|---|---|---|---|---|---|---|---|
| Guibaud-Vainsel syndrom (RTA 3) | ↓ | Acetazolamid | CA2 | CA II | Proximal tubulus | ||
| Renal glykosuri | – | SGLT2-hæmmerea (Canagliflozin, Tofogliflozin, Dapagliflozin) | SLC5A2 | Sodium/glukose cotransporter 2 (SGLT2) | Proximal tubulus | ||
| Bartters syndrom | ↓ | Furosemid | SLC12A1b | Na-K-2Cl cotransporter (NKCC2) | Loop of Henle | ||
| Gitelman syndrom | ↓ | Thiazid | SLC12A3 | Thiazid-sensitiv Na-Cl cotransporter | Distal tubulus | ||
| Diabetes insipidus | – | – | Vaptan | AVPR2, AQP2 | V2-receptor | Kollektorkanalen | |
| Pseudohypoaldosteronizm, AD | ← | Spironolacton | NR3C2 | Spironolacton-sensitiv mineralokortikoidreceptor | Collecting duct | ||
| Pseudohypoaldosteronizm, AR | Amilorid | SCNN1A, SCNNN1B, SCNNN1G | Amiloridfølsom epithelial natriumkanal | Collecting duct | |||
AR – autosomalt recessivt; AD – autosomal dominant.
SGLT2-hæmmere (glukuretika) er de nye lægemidler til behandling af type 2-diabetes; under behandlingen observeres osmotiske diurese-relaterede hændelser såsom pollakiuri og polyuri.
Selv om der findes forskellige Bartter-gener, er dette det gen, der efterligner furosemid.
Angang til hypokaliæmi
GS er også kendt som familiær hypokaliæmisk hypomagnesiæmi, fordi hypokaliæmi er det mest almindelige fænomen. Hos en patient med hypokaliæmi og mistanke om tubulopati bør man bekræfte hyperkaliuri. Renal kaliumsvind kan påvises ved at beregne TKKG, FeK, tilfældigt K/Cr-forhold eller efter 24-timers urinopsamling (tabel 1). Vurdering af kaliuriuri bør foretages, når patienten hverken tager diuretika eller kaliumtilskud, og når kaliumniveauet er lavt. Når kaliumudskillelsen er under 30 mmol/l og TTKG er lav, er hypokaliæmi forårsaget af extrarenalt tab eller et transcellulært K+-shift.
Når overdrevent kaliumrenalt tab hos en patient med hypokaliæmi er bekræftet, baseres den videre diagnose på pH og blodtryk. I mange tilfælde vil disse enkle undersøgelser være nok til at stille diagnosen. Et vigtigt problem af stigende interesse er primær aldosteronisme (PA). Nogle kliniske undersøgelser har antydet, at PA er årsag til over 10 % af arteriel hypertension (AH) og er mere almindelig hos patienter med AH, der er resistente over for antihypertensive midler.19 Hypokaliæmi er et af de “klassiske” symptomer på PA, men er ikke så almindeligt, som det blev anset.20 Men da der ikke findes nogen guldstandard i diagnosen af AP, er det værd at huske på RAA-aksen hos patienter med AH og hypokaliæmi.19
Diagnose af hypomagnesiæmi
Hypomagnesiæmi er en almindelig abnormitet hos GS-patienter, selv om den ikke observeres i alle tilfælde.21 Hos normomagnesiæmiske patienter med GS er den kliniske manifestation og elektrolytafvigelserne mildere.22
Som kaliumioner filtreres magnesiumioner frit af glomeruli. 10 % af det filtrerede Mg2+ absorberes af den proximale tubulus, 50-70 % i den opstigende del af Henles løkke. Distal reabsorption afhænger af epitheliale Mg2+ TRPM6-kanaler. Magnesiuri afhænger af det orale indtag, og FeMg bør beregnes for at fastslå renal Mg2+-spild (FeMg=(urinmagnesium×serumkreatinin)/×100). FeMg under 2 % tyder på dårlig indtagelse, GI-tab eller overførsel af Mg til cellerne. FeMg på over 4 % tyder på renal Mg2+ svind. Når renalt Mg-svind er påvist, bør der beregnes et tilfældigt Ca/Cr-forhold. Urin Ca/Cr-ratio 0,3 er typisk ved hypermagesuri ved Bartters syndrom, det samme er isoleret recessiv hypomagnesiæmi med normocalciuri, familiær hypomagnesiæmi med hypercalciuri og nefrocalcinose, autosomal dominant hypocalcæmi med hypercalciuri, behandling med loopdiuretika og nefropati forårsaget af nogle nefrotoksiner23 .
Magnesium- og kaliumhomøostase er relateret til hinanden, og kaliumdepletion kan ikke korrigeres før korrektion af hypomagnesiæmi.23 I gennemsnit bør patienterne få op til 500mEq kalium, 4-5mg/kg/dag af 5-10mg magnesiumchlorid. Amilorid (5-10mg/dag) og spironolacton (200-300mg) er nyttige.3
Kliniske symptomer og laboratorieresultater er afgørende for diagnosen. I tilfælde med typisk fænotype anbefaler nogle forfattere at udføre en thiazidtest for at bekræfte diagnosen.24 I det forelagte tilfælde afslørede genetisk undersøgelse to nye mutationer i SLC12A3-genet. De er sandsynligvis forårsaget af manglende funktionelle mutationer af NCCT.
Konklusioner
GS syndrom er en af de sjældne årsager til hypokaliæmi, og det synes at være en udfordring for lægerne. Vi viser i denne artikel, at hvis man husker på en meget simpel tilgang til hypokaliæmi og er opmærksom på diuretika virkning og deres lighed med arvelige tubulopatier, kan diagnosen være ret ligetil. GS bør differentieres fra andre tubulopatier (arvelige såvel som erhvervede) og andre årsager til hypokaliæmi (f.eks. Conn’s sygdom). Familiær anamnese kan afsløre asymptomatiske patienter med GS. Passende behandling beskytter patienterne mod potentielt farlige komplikationer.