
En kort introduktion til ChIP-Seq

Protein-DNA-interaktioner anvendes i vid udstrækning til at klarlægge de mekanismer, der ligger til grund for cellens fysiologi. Udviklingen af kromatinimmunopræcipitation (ChIP)-assayteknologien gjorde det muligt at undersøge sådanne mekanismer. Efter yderligere udvikling opstod et alternativ til dyb sekventering (ChiP-Seq), som giver fordele med hensyn til specificitet og følsomhed.
Et ChIP-Seq-forsøg begynder med en tværbinding af hele celler med formaldehyd, efterfulgt af sonicering og DNA-isolering. Derefter udføres immunudfældning af DNA-proteinkomplekset, som består af antistoffer, der binder sig til specifikke proteiner. De dannede immunokomplekser udfældes og oprenses. Endelig sekventeres DNA’et, hvorved der genereres højopløselige data om de berigede steder. Denne fremgangsmåde giver sammen med en veletableret ChIP-seq-pipeline forskerne mulighed for at indfange DNA-transskriptionsfaktorer, histonmodifikationssteder, epigenetiske ændringer og signaturer for genreguleringsnetværk.
Klinisk relevans og anvendelser
Epigenetiske ubalancer på tværs af sygdoms- og sundhedstilstande kan involvere histonmodifikation og ændrede transkriptionsfaktorer. Heri er ChIP-Seq-undersøgelser blevet anvendt til at klarlægge patologiske molekylære mekanismer, der ligger til grund for kræft og andre sygdomme. ChIP-seq-analyser bidrager også til forståelsen af transkriptionsfaktorers rolle i forbindelse med sygdomme. Faktisk synes nogle transkripter at være ændret under manifestationer af kliniske fænotyper.
Oversigt over ChIP-Seq-pipeline
ChIP-Seq-analysepipeline er hovedkomponenten i DNA-proteininteraktionsprojekter og består af flere trin, herunder rå databehandling, kvalitetskontrolanalyse, tilpasning til referencegenomet, kvalitetskontrol af de tilpassede læsninger, peak calling, annotering og visualisering. Det er imidlertid afgørende at have et gennemtænkt eksperimentelt design for at opnå resultater af høj kvalitet i et ChIP-seq-eksperiment. Før du påbegynder din analyse, er det vigtigt at overveje parametre såsom prøve replikater, kontrolgrupper, sekventeringskits og sekventeringsplatforme.
Kvalitetskontrol
Alle Basepair-rapporter indeholder kvalitetsscorer for at hjælpe med at afdække potentielle sekventeringsproblemer eller kontaminering i dine inputdata.
Kvalitetskontrol (QC)-trinnet har til formål at evaluere kvaliteten af high throughput-data genereret fra sekventering. Dette trin svarer til dem, der udføres i DNA-seq- og RNA-seq-analyser. Her omfatter de vigtigste metrikker, der evalueres, sekvens- og basekvalitet, GC-indhold, tilstedeværelse af sekventeringsadaptorer og overrepræsenterede sekvenser. Et af de mest almindeligt anvendte programmer til denne type analyse er FastQC. Hvis der identificeres sekvenser af lav kvalitet, kan de desuden senere fjernes under trimningstrinnet. Selv om det er et valgfrit trin, forbedrer trimning datakvaliteten ved kun at beholde læsninger af høj kvalitet.
Alignment
Efter QC-måling af ChIP-Seq-læsninger justeres de til et referencegenom. Ved hjælp af læsemapping kan forskerne identificere oprindelsen af en læsesekvens i genomet. Populære alignment softwareværktøjer, der anvendes, omfatter Bowtie og BWA, som begge anvendes i Basepairs ChIP-seq-pipelines. Begge værktøjer kortlægger lavt divergerende sekvenser i forhold til et referencegenom.
Læsningstællingsflowet hjælper med at give et overblik over det store billede af brugbare læsninger ved afslutningen af trimnings-, tilpasnings- og deduplikeringsprocesserne. Tænk på figuren som et samlebånd til dataanalyse: indtast rå data, få et output af anvendelige læsninger.
Kvalitetskontrol af de justerede læsninger
Det næste trin består af QC-inferens af det justerede datasæt. Under kortlægningsprocessen forårsager læseduplikater, der indføres ved PCR-amplifikation og sekventering, bias under peak calling- og berigelsesanalysen. Basepair anvender Picard-værktøjet til at fjerne duplikater. Når duplikater er blevet fjernet, bør du evaluere Non-Redundant Fraction (NRF) af de justerede læsninger. NRF måler de unikke læsninger, der mappes til referencegenomet. Ideelle ChIP-seq-eksperimenter bør have mindre end tre reads pr. position.
Peak Calling
Peak Calling-trinnet registrerer berigede protein-DNA-interaktionsregioner på genomet. Basepair’s ChIP-seq-pipeline bruger MACS2 til at udføre denne analyse. I MACS2 udføres peak calling på grundlag af tre hovedtrin: fragmentestimation, efterfulgt af identifikation af lokale støjparametre og derefter peakidentifikation. Som resultat af dette trin får brugerne en endelig tabel med oplysninger om toppe, f.eks. berigelsesscore, -log10p-værdi, -log10q-værdi og positionen til start af toppe. Det kan stærkt anbefales at anvende kontrolprøver i dette trin til sammenligning med det undersøgte måldatasæt. Husk på, at gode kontrolgrupper giver mere pålidelige resultater.
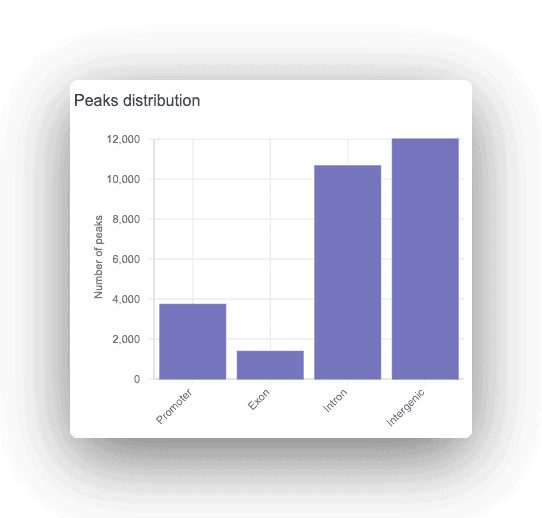
Hvert peak annoteres som promotor, intronisk eller intergenisk, og det tilsvarende gen vises. For alle fundne toppe udføres en motivanalyse for at finde overrepræsenterede transkriptionsfaktorbindingssteder.
Oversigt over resultater
En ChIP-seq-pipeline kan ikke kun give oplysninger om kromatintilstanden, men også om transkriptionsfaktorbinding i en bestemt gen- eller loci-kontekst. Forekomsten af histonmodifikationer og transkriptionsfaktorer i DNA-regulerende regioner kan udgøre en tilstandsspecifik epigenetisk signatur. Epigenetiske forstyrrelser kan således være forbundet med kliniske fænotyper. F.eks. kan heterogenitet i kromatintilstande føre til behandlingsresistens ved brystkræft. Disse celler har tendens til at miste repressive histonmodifikationsmarkører og yderligere øge ekspressionen af gener, der er kendt for at fremme resistens over for kræftbehandling.
Peak, Motif og Pathway Analysis in ChIP-Seq Analysis Pipeline
Identifikation af motivtranskriptionsfaktorberigelse anvendes til at belyse, om transkriptionsfaktorer samarbejder eller konkurrerer i et givet område. Identifikation af toppe i DNA-motivregioner kan forbedre fortolkningen af de eksperimentelle resultater. Tilsammen giver både peak- og motivanalyser indsigt i, hvad der kan foregå i en celle. Integrationen af peak- og motivberigelser resulterer i et epigenomisk landskab med mulige biologiske konsekvenser. Desuden bruges pathway-analyser til at identificere proteiner i en pathway. Undersøgelser og konklusioner formuleres på baggrund af en tilstedeværelse af et protein.
Datovisualisering
Resultatdata fra en ChIP-seq-pipeline kan visualiseres ved hjælp af en genom-browser. Basepair-rapporter indeholder en indlejret IGV2-genombrowser, der giver dig mulighed for at interagere med dine data. Data kan alternativt visualiseres ved hjælp af heatmaps, som er repræsentative intensitetsinfografikker baseret på datatæthed, der viser tilstedeværelsen eller fraværet af specifikke mærker. Andre grafikker, der anvendes her, omfatter berigelsesplot, upSet og dækningsplot, som både beregner og viser dækningen af peak-regioner over genomet.
Genombrowseren er et fantastisk værktøj til at visualisere dine rå genomiske data. Det er indbygget i hver ChIP-seq-analyserapport på Basepair.
1. Grosselin, K., A. Durand, et al. High-throughput single-cell ChIP-seq identificerer heterogenitet af kromatintilstande i brystkræft. Nat Genet, v.51, n.6, jun, s.1060-1066. 2019.
2. Northrup, D. L. e K. Zhao. Anvendelse af ChIP-Seq og beslægtede teknikker til undersøgelse af immunfunktion. Immunity, v.34, n.6, jun 24, s.830-42. 2011.
3. Park, S. J., J. H. Kim, et al. A ChIP-Seq Data Analysis Pipeline Based on Bioconductor Packages. Genomics Inform, v.15, n.1, marts, s.11-18. 2017.
4. Pepke, S., B. Wold, et al. Computation for ChIP-seq and RNA-seq studies. Nat Methods, v.6, n.11 Suppl, Nov, s.S22-32. 2009.
5. Satoh, J., N. Kawana, et al. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul Syst Bio, v.7, s.139-52. 2013.