
Introduktion
Hypokalemi är ett vanligt kliniskt problem hos endokrinologer och nefrologer. Det finns många uppenbara orsaker till hypokalemi såsom diarré, kräkningar eller missbruk av diuretika. Andra orsaker som tubulopatier observeras sällan och deras diagnos är mer utmanande. Det finns många ärftliga och förvärvade tubulopatier som orsakar hypokalemi, ibland allvarlig och livshotande.1
En relativt vanlig men förbisedd orsak till hypokalemi är Gitelmans syndrom (GS).2 Det är en recessiv saltförlorande tubulopati som orsakas av SLC12A3-genmutationen. SLC12A3-genen kodar för den tiazidkänsliga transportören NCCT (sodium chloride co-transporter). NCCT finns i de distala konvoluterade tubulära cellerna (DCC), som ansvarar för 7-10 % av den tubulära absorptionen av elektrolyter.3
De allvarligaste laboratorieavvikelserna som hittas vid GS är hypokalemi och hypomagnesemi som orsakas av renal K+- och Mg2+-förlust. Andra typiska förändringar är metabolisk alkalos, hypokalciuri och hyperreninemisk hyperaldosteronism.4 Mild till måttlig hypofosfatemi observeras ofta.5 Allvarlig hypofosfatemi med svår hyponatremi har också rapporterats.6,7
De första symtomen på GS uppträder hos barn eller unga vuxna med normal tillväxt och historia av saltkrävande beteenden (barn som gärna äter gurka eller saltlake, saltade gurkor, apelsiner och citroner, barn som slickar salt från potatischips etc.).8 Den kliniska presentationen varierar mellan olika patienter. Vissa är symtomfria men andra utvecklar livshotande komplikationer. Män manifesterar en allvarligare fenotyp än kvinnor.8 De vanligaste symtomen är muskelkramper och svaghet, förstoppning, nokturi, polyuri, törst, polydipsi, hjärtarytmier, parestesier och ökad salthunger. Arteriell hypotension är vanligt och i många fall det mest framträdande symtomet, men i den åldrande GS-populationen kan hypertoni förekomma.8 Korrelationen mellan biokemiska avvikelser och symtom är inte stark.9 GS stör inte barnens humör och sociala relationer.9,10 I övrigt är symtom vanligare hos vuxna och kan ha negativ inverkan på deras livskvalitet. Fyrtiofem procent av GS-patienterna anser att deras symtom är ett måttligt till stort problem.11 Extrem utmattning, muskelsvaghet, parestesier, allvarlig trötthet och hypotoni är förknippade med mild till allvarlig minskning av dagliga aktiviteter.9
Den uppskattade prevalensen av GS är 1:40 0008 och prevalensen av heterozygot är minst 1 % i den europeiska befolkningen. Mer än 180 olika mutationer i SLC12A3 har beskrivits hittills.12
Fallbeskrivning
En 26-årig man lades in på sjukhus på grund av förekomst av synkope, generaliserad och muskelsvaghet och muskelkramper. Patientens anamnes avslöjade en episod av synkope med en kaliumnivå på 3,16 mmol/l. Vid ytterligare uppföljning i öppenvårdsbedömning observerades återkommande förekomst av hypokalemi (det lägsta värdet 2,6mmol/l). Blodtrycket var normalt 110/80, hjärtfrekvensen var 72 per minut; det fanns inga förändringar i den fysiska undersökningen. Inga neurologiska fynd, vikt 74 kg, längd 178 cm. Vid intagningen på sjukhuset visade laboratorieutredningen mild hypokalemi (K+ 3,0mmol/l), hypomagnesemi (Mg2+ 1,36mg/dl), hypokalciuri (
mg/24h) och metabolisk alkalos (HCO3- 29,7mmol/l, BE 5,3mmol/l). Njurfunktionen var god med eGFR>60ml/min. Bilddiagnostiska undersökningar var okej, likaså EKG.
Fortsatta undersökningar bekräftade att hypokalemi orsakades av renal kaliumförlust. Kaliumförlust över 24 timmar, transtubulär K+ gradient (TTKG), fraktionell K+ utsöndring (FeK) och slumpmässig K/kreatinin kvot (K/Cr) var typiska för hyperkaliuri och visas i tabell 1.13-15 Daglig magnesiuri var 64 mg/24h, med en fraktionell magnesiumutsöndring (FeMg) på 15 %.
Värdena är diagnostiska för hypokalemi.13-15
| Värden som diagnostiserar olämplig kaliures | Patient | ||
|---|---|---|---|
| Urinär K+ | >20mmol/24h Om serum K+3.5mmol/l |
57mmol/l (serum K+=3.0mmol/l) |
|
| FeK=×100% | >6,5% Om serum K+3.5mmol/l |
12% (serum K+=3.0mmol/l) |
|
| TTKG= | TTKG>2 Om serum K+3.5mmol/l |
7,3 (serum K+=3,0mmol/l) |
|
| uK/Cr – slumpmässigt K+ i urinen i förhållande till kreatinin | uK/Cr>15mmol/g Om serum K+3.5mmol/l |
67mmol/g (serum K+=3.0mmol/l) |
|
Trots att blodtrycket var normalt utfördes hormonella tester för att utesluta Conn’s sjukdom och CT av buken, som inte visade på några avvikelser i binjurarna. Sekundär hyperaldosteronism med nivåer av aldosteron 289pg/ml (normalintervall 20-180) och renin 205mIU/ml (normalintervall 2,8-39,9) var typiska för GS.
Patienten diagnostiserades som GS utifrån en klinisk fenotyp. Efter diagnosen inleddes behandling med tillskott av kalium och magnesium. Han fick 20 mmol kaliumklorid och 18 mmol magnesiumpyrrolidonkarboxylat, med klinisk förbättring. Familjeanamnesen avslöjade mild asymtomatisk hypokalemi hos patientens 35-åriga syster. Men undersökningar visade varken hyperkaliuri eller hypomagnesemi. GS diagnostiserades inte i detta fall.
Genetisk analys
Enligt den genetiska analysen, algoritm föreslagen av Nozu16SLC12A3-genen bör testas hos patienter med hypokaliemässig metabolisk alkalos, med fullgången födsel, normalvikt utan nefrokalcinos, med hypokaluria och hypomagnesemi.
I det beskrivna fallet utfördes genetisk testning och två heterozygota mutationer: c.35_36insA och c.1095+5G>A hittades i transkript NM_000339.2 av SLC12A3-genen (Fig. 1). Båda mutationerna finns ännu inte i den tillgängliga HGMD(r)-databasversionen . Den första mutationen hittades också hos patientens mor och den andra hos fadern. Endast en av de två mutationer som identifierades hos vår patient c.35_36insA hittades hos hans syster.
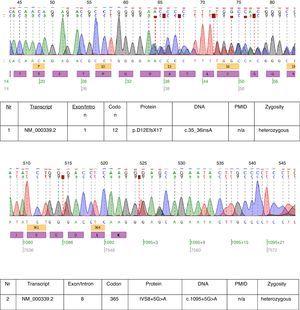
Kromatogram av den drabbade patientens DNA-sekvensering.
De mutationer som hittades hos den här patienten bevisar nästan den kliniska diagnosen GS. Den patogenetiska betydelsen av ramförskjutningsmutationen är uppenbar. Vi kan inte vara säkra på den patogenetiska relevansen av mutationen på spliceplatsen i intron 8. Konsensussplickssekvensen G (100 %), T (100 %), A (62 %), A (68 %), G (82 %), T (63 %) vid splicksdonatorn i intron 8 stämmer inte exakt överens med konsensus GTgAGc. Den mutation som hittades hos denna patient avviker ytterligare från konsensussekvensen (GtgAac). Också in silico-analysen med mutationstester anser att denna mutation är sjukdomsorsakande.17
Diskussion
Tubulopatier är sällsynta sjukdomar. Enligt databasen RenalTube18 är de vanligaste primära tubulopatierna i den europeiska befolkningen distal renal tubulär acidos, Bartters syndrom, familjär hypomagnesemi med hyperkalciuri och GS.18 Prevalensen av GS är cirka 25 fall per 1 miljon.3 Eftersom GS är en av de vanligaste kommer troligen många nefrologer och endokrinologer att konfronteras med fall av GS under sin karriär. Vår patient hade en typisk klinisk presentation av GS och svarade väl på behandlingen. Problemet med GS är att förbisedd hypokalemi kan orsaka dödsfall på grund av hjärtstillestånd eller förlamning av andningsmusklerna. Det allvarliga neuromuskulära symtomet såsom hypokalemisk förlamning förekommer hos upp till 6 % av patienterna (vanligare hos asiatiska patienter).3
De ärftliga tubulopatierna har ett slags ”spegelbilder”-förvärvade tubulopatier orsakade av diuretika. Alla diuretika (för att vara exakt: natriuretika, acquaretika eller glukuretika) orsakar abnormiteter som liknar dem som finns i ärftliga tubulopatier (tabell 2). Nästan i alla tubulopatier förekommer polyuri, och förändringar i kaliumnivån hör till de vanligaste problemen. Hos patienter med GS hittas mutationer i SLC12A3-genen. Denna gen kodar för den thiazidkänsliga transportören. Därför liknar GS kronisk behandling med tiazider.
Diuretika och tubulopatier.
| Ärftlig enkel genfel | Kaliumkoncentration | Symtom som liknar användning av: | Gen | Genprodukt | Genprodukts placering | |
|---|---|---|---|---|---|---|
| Guibaud-Vainsel syndrom (RTA 3) | ↓ | Acetazolamid | CA2 | CA II | Proximal tubuli | |
| Renal glykosuri | – | SGLT2-hämmarea (canagliflozin, Tofogliflozin, Dapagliflozin) | SLC5A2 | Natrium/glukos cotransporter 2 (SGLT2) | Proximal tubulus | |
| Bartters syndrom | ↓ | Furosemid | SLC12A1b | Na-K-2Cl-kotransporter (NKCC2) | Henleslussen | |
| Gitelmans syndrom | ↓ | Thiazid | SLC12A3 | Thiazid-känslig Na-Cl cotransporter | Distal tubulus | |
| Diabetes insipidus | – | – | Vaptan | AVPR2, AQP2 | V2-receptor | Samlingsrör |
| Pseudohypoaldosteronizm, AD | ← | Spironolakton | NR3C2 | Spironolakton-känslig mineralokortikoidreceptor | Collecting duct | |
| Pseudohypoaldosteronizm, AR | Amilorid | SCNN1A, SCNNN1B, SCNNN1G | Amiloridkänslig epitelial natriumkanal | Collecting duct | ||
AR – autosomal recessiv; AD – autosomalt dominant.
SGLT2-hämmare (glukuretika) är de nya läkemedlen för behandling av typ 2-diabetes; under behandlingen observeras osmotiska diureserelaterade händelser som pollakiuri och polyuri.
Och även om det finns olika Barttergener är detta den som efterliknar furosemid.
Ansats på hypokalemi
GS kallas också för familjär hypokaliemisk hypomagnesemi, eftersom hypokalemi är det vanligaste fenomenet. Hos en patient med hypokalemi och misstanke om tubulopati bör man bekräfta hyperkaliuri. Renal kaliumförlust kan bevisas genom beräkning av TKKG, FeK, slumpmässig K/Cr-kvot eller efter 24-timmars urinprovtagning (tabell 1). Bedömning av kaliuriuri bör göras när patienten varken tar diuretika eller kaliumtillskott och när kaliumnivån är låg. När kaliumutsöndringen är under 30 mmol/l och TTKG är låg orsakas hypokalemi av extrarenal förlust eller en transcellulär K+-förskjutning.
När överdriven kaliumförlust från njurarna hos en patient med hypokalemi bekräftas, baseras den fortsatta diagnosen på pH och blodtryck. I många fall räcker dessa enkla undersökningar för att ställa diagnosen. Ett viktigt problem av ökande intresse är primär aldosteronism (PA). Vissa kliniska studier har föreslagit att PA är orsaken till över 10 % av arteriell hypertoni (AH) och är vanligare hos patienter med AH som är resistenta mot antihypertensiva medel.19 Hypokalemi är ett av de ”klassiska” symptomen på PA, men det är inte så vanligt som man trodde.20 Men eftersom det inte finns någon guldstandard för diagnosen av AP är det ändå värt att komma ihåg RAA-axeln hos patienter med AH och hypokalemi.19
Diagnos av hypomagnesemi
Hypomagnesemi är en vanlig avvikelse hos GS-patienter, även om den inte observeras i alla fall.21 Hos normomagnesemiska patienter med GS är den kliniska manifestationen och elektrolytavvikelserna mildare.22
Som kaliumjoner filtreras magnesiumjoner fritt av glomeruli. 10 % av det filtrerade Mg2+ absorberas av den proximala tubuli, 50-70 % i den uppåtgående delen av Henles slinga. Distal reabsorption beror på epiteliala Mg2+ TRPM6-kanaler. Magnesiuri beror på oralt intag och FeMg bör beräknas för att fastställa renal Mg2+-förlust (FeMg=(urinmagnesium×serumkreatinin)/×100). FeMg mindre än 2 % tyder på dåligt intag, gastrointestinala förluster eller förflyttning av Mg till cellerna. FeMg över 4 % tyder på renal Mg2+-förlust. När renal Mg-försämring är bevisad bör slumpmässigt Ca/Cr-förhållandet beräknas. Urinens Ca/Cr-kvot 0,3 är typisk för hypermagesuri vid Bartters syndrom, liksom isolerad recessiv hypomagnesemi med normokalciuri, familjär hypomagnesemi med hyperkalciuri och nefrokalcinos, autosomal dominant hypokalcemi med hyperkalciuri, behandling med loopdiuretika och nefropati orsakad av vissa nefrotoxiner.23
Magnesium- och kaliumhomeostas är relaterade till varandra, och kaliumdepletion kan inte korrigeras förrän hypomagnesemi har korrigerats.23 I genomsnitt bör patienterna få upp till 500mEq kalium, 4-5mg/kg/dag av 5-10mg magnesiumklorid. Amilorid (5-10 mg/dag) och spironolakton (200-300 mg) är till hjälp.3
Kliniska symtom och laboratorieresultat är viktiga för diagnosen. I fall med typisk fenotyp rekommenderar vissa författare att man utför ett tiazidtest för att bekräfta diagnosen.24 I det presenterade fallet avslöjade den genetiska undersökningen två nya mutationer i SLC12A3-genen. De orsakas troligen av brist på funktionella mutationer av NCCT.
Slutsatser
GS syndrom är en av de sällsynta orsakerna till hypokalemi, och det verkar vara en utmaning för läkare. Vi visar i denna artikel att om man minns om ett mycket enkelt tillvägagångssätt vid hypokalemi och är medveten om diuretikas verkan och deras likhet med ärftliga tubulopatier kan diagnosen vara ganska okomplicerad. GS bör särskiljas från andra tubulopatier (ärftliga såväl som förvärvade) och andra orsaker till hypokalemi (t.ex. Conn’s sjukdom). Familjeanamnesen kan avslöja asymtomatiska patienter med GS. Lämplig behandling skyddar patienterna från potentiellt farliga komplikationer.