
Lyhyt johdatus ChIP-Seq-analyysiin

Valkuaisen ja DNA:n välisiä vuorovaikutussuhteita hyödynnetään laajalti solun fysiologian taustalla vaikuttavien mekanismien selvittämiseksi. Kromatiinin immunoprecipitointitekniikan (ChIP) kehittäminen mahdollisti tällaisten mekanismien tutkimisen. Jatkokehityksen jälkeen syntyi syväsekvensointivaihtoehto (ChiP-Seq), joka tarjoaa etuja spesifisyyden ja herkkyyden suhteen.
ChIP-Seq-koe alkaa koko solun ristisidonnalla formaldehydillä, jota seuraa sonikointi ja DNA:n eristäminen. Tämän jälkeen suoritetaan DNA-proteiinikompleksin immunoprecipitaatio, joka koostuu tiettyihin proteiineihin sitoutuvista vasta-aineista. Muodostuneet immunokompleksit saostetaan ja puhdistetaan. Lopuksi DNA sekvensoidaan, jolloin saadaan korkearesoluutioiset tiedot rikastuneista kohdista. Tämä lähestymistapa yhdessä vakiintuneen ChIP-seq-putken kanssa antaa tutkijoille mahdollisuuden kaapata DNA:n transkriptiotekijöitä, histonimodifikaatiokohtia, epigeneettisiä muutoksia ja geenien säätelyverkostojen allekirjoituksia.
Kliininen merkitys ja sovellukset
Epigeneettiseen epätasapainoon sairauksissa ja terveystiloissa voi liittyä histonimodifikaatioita ja muuttuneita transkriptiotekijöitä. Tässä yhteydessä ChIP-Seq-tutkimuksia on käytetty syövän ja muiden sairauksien taustalla olevien patologisten molekyylimekanismien selvittämiseen. ChIP-seq-analyysit edistävät myös transkriptiotekijöiden roolin ymmärtämistä sairauksien aikana. Itse asiassa jotkin transkriptiot näyttävät muuttuvan kliinisen fenotyypin ilmenemisen aikana.
Katsaus ChIP-Seq-analyysiputkeen
ChIP-Seq-analyysiputki on DNA-proteiinien vuorovaikutusprojektien pääkomponentti, ja se koostuu useista vaiheista, kuten raakadatan käsittelystä, laadunvalvonta-analyyseistä, kohdentamisesta referenssigenomiin, kohdentuneiden lukulukemien laaduntarkistuksesta, piikkien kutsumisesta, annotoinnista ja visualisoinnista. Harkittu koesuunnitelma on kuitenkin ratkaisevan tärkeää laadukkaiden tulosten saamiseksi ChIP-seq-kokeessa. Ennen analyysin aloittamista on olennaista ottaa huomioon parametrit, kuten näytereplikaatit, kontrolliryhmät, sekvensointipaketit ja sekvensointialustat.
Laadunvalvonta
Kaikissa Basepair-raporteissa on laatupisteet, joiden avulla voidaan paljastaa mahdolliset sekvensointiin liittyvät ongelmat tai kontaminaatio syöttämässäsi datassa.
Laadunvalvonnan (Quality Control, QC) vaiheen tavoitteena on arvioida sekvensoinnilla tuotetun suurten läpimenoaikojen datan laatua. Tämä vaihe on samanlainen kuin DNA-seq- ja RNA-seq-analyyseissä. Tässä arvioitavia päämittareita ovat sekvenssin ja emäksen laatu, GC-pitoisuus, sekvensointiadapterien esiintyminen ja yliedustetut sekvenssit. Yksi tämäntyyppiseen analyysiin yleisimmin käytetyistä ohjelmista on FastQC. Jos huonolaatuisia sekvenssejä tunnistetaan, ne voidaan myöhemmin poistaa trimmausvaiheessa. Vaikka trimmaus on valinnainen vaihe, se parantaa datan laatua säilyttämällä vain korkealaatuisia lukuja.
Alignment
QC-mittauksen jälkeen ChIP-Seq-lukemat kohdistetaan referenssigenomiin. Lukukartoituksen avulla tutkijat voivat tunnistaa lukusekvenssin alkuperän genomissa. Suosittuja käytettyjä kohdistamisohjelmistoja ovat Bowtie ja BWA, joita molempia käytetään Basepairin ChIP-seq-pipeleissä. Molemmat työkalut kartoittavat vähän poikkeavia sekvenssejä referenssigenomia vasten.
Lukulukuvirta auttaa antamaan yleiskuvan käyttökelpoisista lukusarjoista trimmaus-, kohdistamis- ja deduplikointiprosessien päätteeksi. Ajattele kuviota data-analyysin liukuhihnana: syötät raakadataa, saat tulosteeksi käyttökelpoiset lukemat.
Alignoitujen lukemien laaduntarkastus
Seuraava vaihe koostuu alignoidun tietokokonaisuuden laaduntarkkailusta. Kartoitusprosessin aikana PCR-monistamisen ja sekvensoinnin aiheuttamat lukuduplikaatit aiheuttavat harhaa piikkien kutsumisessa ja rikastusanalyysissä. Basepair käyttää Picard-työkalua duplikaattien poistamiseen. Kun duplikaatit on poistettu, kannattaa arvioida kohdistettujen lukujen ei-redundanttifraktio (NRF). NRF mittaa referenssigenomia vastaavien lukujen ainutlaatuisuutta. Ihanteellisissa ChIP-seq-kokeissa pitäisi olla alle kolme lukua per positio.
Peak Calling
Peak Calling -vaiheessa havaitaan rikastuneet proteiini-DNA-vuorovaikutusalueet genomissa. Basepairin ChIP-seq-putki käyttää MACS2:ta tämän analyysin suorittamiseen. MACS2:ssa piikkien kutsuminen perustuu kolmeen päävaiheeseen: fragmenttien estimointiin, jota seuraa paikallisten kohinaparametrien tunnistaminen ja sitten piikkien tunnistaminen. Tämän vaiheen tuloksena käyttäjät saavat lopullisen taulukon, jossa on piikkitietoja, kuten rikastumispisteet, -log10p-arvo, -log10q-arvo ja sijainti piikin alkuun nähden. Tässä vaiheessa on erittäin suositeltavaa käyttää kontrollinäytteitä, jotta niitä voidaan verrata tutkittuun kohdetietoaineistoon. Pidä mielessä, että hyvät kontrolliryhmät tuovat luotettavampia tuloksia.
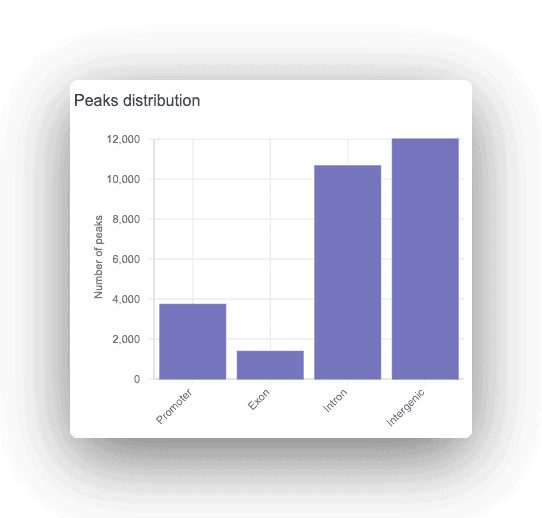
Jokainen piikki on annotoitu promoottoriksi, introniseksi tai intergeeniseksi, ja vastaava geeni näytetään. Kaikille löydetyille piikeille tehdään motiivianalyysi yliedustettujen transkriptiotekijöiden sitoutumiskohtien löytämiseksi.
Tulosten yleiskuvaus
ChIP-seq-putkilinja voi antaa tietoa kromatiinin tilan lisäksi myös transkriptiotekijöiden sitoutumisesta määritellyn geenin tai lokusten kontekstissa. Histonimodifikaatioiden ja transkriptiotekijöiden esiintyminen DNA:n säätelyalueilla voi muodostaa tilakohtaisen epigeneettisen allekirjoituksen. Näin epigeneettiset häiriöt voidaan liittää kliinisiin fenotyyppeihin. Esimerkiksi kromatiinitilojen heterogeenisuus voi johtaa hoitoresistenssiin rintasyövässä. Näillä soluilla on taipumus menettää repressiivisiä histonimodifikaatioiden markkereita ja lisätä edelleen sellaisten geenien ilmentymistä, joiden tiedetään edistävän resistenssiä syöpähoitoa vastaan.
Peak-, motiivi- ja polkuanalyysi ChIP-Seq-analyysiputkistossa
Transkriptiotekijöiden rikastumismotiivien tunnistamista käytetään selvittämään, ovatko transkriptiotekijät yhteistyöhön osallistuvia vai keskenään kilpailevia tietyllä alueella. Huippujen tunnistaminen DNA-motiivialueilla voi parantaa kokeellisten tulosten tulkintaa. Yhdessä sekä piikki- että motiivianalyysit antavat tietoa siitä, mitä solussa saattaa tapahtua. Huippu- ja motiivirikastumien yhdistäminen johtaa epigenomiseen maisemaan, jolla voi olla biologisia seurauksia. Lisäksi polkuanalyysiä käytetään polun proteiinien tunnistamiseen. Tutkimukset ja johtopäätökset muotoillaan proteiinien läsnäolon perusteella.
Datan visualisointi
ChIP-seq-putken tulokset voidaan visualisoida genomiselaimen avulla. Basepair-raportit sisältävät upotetun IGV2-genomiselaimen, jonka avulla voit olla vuorovaikutuksessa datasi kanssa. Tiedot voidaan vaihtoehtoisesti visualisoida heatmapeilla, jotka ovat datatiheyteen perustuvia edustavia intensiteetti-infografioita, jotka osoittavat tiettyjen merkkien läsnäolon tai puuttumisen. Muita tässä käytettyjä grafiikoita ovat rikastumispiirros, upSet ja kattavuuspiirros, joka sekä laskee että näyttää piikkialueiden kattavuuden koko genomissa.
Genomiselain on loistava työkalu genomin raakadatan visualisointiin. Se on sisällytetty jokaiseen Basepairin ChIP-seq-analyysiraporttiin.
1. Grosselin, K., A. Durand, et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet, v.51, n.6, Jun, s.1060-1066. 2019.
2. Northrup, D. L. e K. Zhao. ChIP-Seqin ja siihen liittyvien tekniikoiden soveltaminen immuunijärjestelmän toiminnan tutkimiseen. Immunity, v.34, n.6, Jun 24, s.830-42. 2011.
3. Park, S. J., J. H. Kim, et al. A ChIP-Seq Data Analysis Pipeline Based on Bioconductor Packages. Genomics Inform, v.15, n.1, Mar, s.11-18. 2017.
4. Pepke, S., B. Wold, et al. Computation for ChIP-seq and RNA-seq studies. Nat Methods, v.6, n.11 Suppl, Nov, s.S22-32. 2009.
5. Satoh, J., N. Kawana, et al. ChIP-Seq-pohjaisten NRF1-kohdegeenien polkuanalyysi viittaa loogiseen hypoteesiin niiden osallistumisesta neurodegeneratiivisten sairauksien patogeneesiin. Gene Regul Syst Bio, v.7, s.139-52. 2013.