
Introduction
Hypokalemia on yleinen kliininen ongelma endokrinologien ja nefrologien käytännössä. Hypokalemiaan on monia ilmeisiä syitä, kuten ripuli, oksentelu tai diureettien väärinkäyttö. Muita syitä, kuten tubulopatioita, havaitaan harvoin, ja niiden diagnosointi on haastavampaa. On olemassa monia perinnöllisiä ja hankittuja tubulopatioita, jotka aiheuttavat hypokalemiaa, joskus vakavaa ja hengenvaarallista.1
Suhteellisen yleinen mutta unohdettu hypokalemian syy on Gitelmanin oireyhtymä (Gitelman syndrome, GS).2 Kyseessä on resessiivinen suolaa menettävä tubulopatia, jonka aiheuttaa SLC12A3-geenimutaatio. SLC12A3-geeni koodaa tiatsidiherkkää transporteria NCCT (natriumkloridi co-transporter). NCCT sijaitsee distaalisissa kierteisissä tubulussoluissa (distal convoluted tubular cells, DCC), jotka vastaavat 7-10 %:sta elektrolyyttien tubulaarisesta imeytymisestä.3
Suureimmat GS:ssä todetut laboratoriopoikkeavuudet ovat hypokalemia ja hypomagnesemia, jotka johtuvat munuaisten K+:n ja Mg2+:n tuhlauksesta. Muita tyypillisiä muutoksia ovat metabolinen alkaloosi, hypokalsiuria ja hyperrenineeminen hyperaldosteronismi.4 Lievää tai keskivaikeaa hypofosfatemiaa havaitaan usein.5 Myös vakavaa hypofosfatemiaa, johon liittyy vaikea hyponatremia, on raportoitu.6,7,7
GS:n ensimmäiset oireet ilmenevät lapsilla tai nuorilla aikuisilla, joiden kasvu on normaali ja joilla on ollut suolaa himoitsevaa käyttäytymistä (lapset syövät innokkaasti suolakurkkua tai suolavettä, suolattuja kurkkuja, appelsiineja ja sitruunoita, lapset nuolevat suolaa perunalastuista jne.8 Kliininen oirekuva vaihtelee potilaiden välillä. Jotkut ovat oireettomia, mutta toisille kehittyy hengenvaarallisia komplikaatioita. Miesten fenotyyppi on vaikeampi kuin naisten.8 Yleisimpiä oireita ovat lihaskrampit ja -heikkous, ummetus, nokturia, polyuria, jano, polydipsia, sydämen rytmihäiriöt, parestesiat ja lisääntynyt suolahalu. Arteriaalinen hypotensio on yleinen ja monissa tapauksissa merkittävin oire, mutta ikääntyvässä GS-väestössä voi esiintyä hypertensiota.8 Biokemiallisten poikkeavuuksien ja oireiden välillä ei ole vahvaa korrelaatiota.9 GS ei vaikuta lasten mielialaan ja sosiaalisiin suhteisiin.9,10 Muut oireet ovat yleisempiä aikuisilla, ja niillä voi olla kielteinen vaikutus heidän elämänlaatuunsa. Neljäkymmentäviisi prosenttia GS-potilaista pitää oireitaan kohtalaisena tai suurena ongelmana.11 Äärimmäiseen uupumukseen, lihasheikkouteen, parestesioihin, vaikeaan väsymykseen ja hypotensioon liittyy päivittäisten toimintojen lievää tai vakavaa vähenemistä.9
GS:n arvioitu esiintyvyys on 1:40 0008 , ja heterotsygoottisen taudin esiintyvyys on vähintään 1 % Euroopan väestössä. SLC12A3:ssa on tähän mennessä kuvattu yli 180 erilaista mutaatiota.12
Tapauskuvaus
26-vuotias mieshenkilö otettiin sairaalaan synkopeen, yleistyneen ja lihasheikkouden sekä lihaskrampien esiintymisen vuoksi. Potilaan anamneesissa ilmeni synkopeekohtaus, jonka kaliumpitoisuus oli 3,16 mmol/l. Avohoidon jatkoseurannassa havaittiin toistuvaa hypokalemiaa (alin arvo 2,6 mmol/l). Verenpaine oli normaali 110/80, syke oli 72 minuutissa; fyysisessä tutkimuksessa ei ollut muutoksia. Ei myöskään neurologisia löydöksiä, paino 74kg, pituus 178cm. Sairaalaan tullessa laboratorioarviossa todettiin lievä hypokalemia (K+ 3,0mmol/l), hypomagnesemia (Mg2+ 1,36mg/dl), hypokalsiuria (
mg/24h) ja metabolinen alkaloosi (HCO3- 29,7mmol/l, BE 5,3mmol/l). Munuaisten toiminta oli hyvä, eGFR>60ml/min. Kuvantamistutkimukset olivat merkitsemättömiä, samoin EKG.
Jatkotutkimukset vahvistivat, että hypokalemia johtui munuaisten kaliuminhukasta. 24 tunnin kaliumhukka, transtubulaarinen K+ -gradientti (TTKG), fraktionaalinen K+ -erittyminen (FeK) ja satunnainen K/kreatiniinisuhde (K/Cr) olivat tyypillisiä hyperkaliurialle, ja ne on esitetty taulukossa 1.13-15 Päivittäinen magnesiuria oli 64 mg/24h, ja fraktionaalinen magnesiumin (FeMg) erittyminen oli 15 %.
Hypokalemian diagnostiset arvot.13-15
| Epäasianmukaisen kaliureesin diagnostiset arvot | Potilas | |
|---|---|---|
| Urinaarinen K+ | >20mmol/24h Jos seerumin K+3.5mmol/l |
57mmol/l (seerumin K+=3.0mmol/l) |
| FeK=×100% | >6.5% Jos seerumin K+3.5mmol/l |
12% (seerumin K+=3.0mmol/l) |
| TTKG= | TTKG>2 Jos seerumin K+3.5mmol/l |
7.3 (seerumin K+=3.0mmol/l) |
| uK/Cr – satunnainen virtsan K+:n ja kreatiniinin välinen suhdeluku | uK/Cr>15mmol/g Jos seerumin K+3.5mmol/l |
67mmol/g (seerumin K+=3.0mmol/l) |
Vaikka verenpaine olikin normaali, tehtiin hormonikokeet Connin taudin poissulkemiseksi ja vatsan tietokonetomografia, eivätkä ne paljastaneet lisämunuaisten poikkeavuuksia. Sekundaarinen hyperaldosteronismi, jonka aldosteronipitoisuudet 289pg/ml (normaali alue 20-180) ja reniini 205mIU/ml (normaali alue 2,8-39,9) olivat GS:lle tyypillisiä.
Potilaalla diagnosoitiin GS kliinisen fenotyypin perusteella. Diagnoosin jälkeen aloitettiin hoito kalium- ja magnesiumlisillä. Hän sai 20 mmol kaliumkloridia ja 18 mmol magnesiumpyrrolidonikarboksylaattia, ja kliininen tilanne parani. Perhehistoriassa todettiin potilaan 35-vuotiaalla siskolla lievä oireeton hypokalemia. Tutkimuksissa ei kuitenkaan ilmennyt hyperkaliuriaa eikä hypomagnesemiaa. GS:ää ei diagnosoitu tässä tapauksessa.
Geneettinen analyysi
Nozun ehdottaman geneettisen analyysin algoritmin mukaan16SLC12A3-geeni tulisi testata potilailla, joilla on hypokalemiaa aiheuttava metabolinen alkaloosi, täysiaikainen syntymä, normaalipainoinen ilman nefrokalsinoosia ja joilla on hypokalsuria ja hypomagnesemia.
Kuvatussa tapauksessa suoritettiin geneettinen testaustutkimus, ja siinä havaittiin kaksi heterotsygoottista mutaatiota: c.35_36insA ja c.1095+5G>A löydettiin SLC12A3-geenin transkriptiosta NM_000339.2 (kuva 1). Molempia mutaatioita ei ole vielä HGMD(r)-tietokannan saatavilla olevassa versiossa . Ensimmäinen mutaatio löytyi myös potilaan äidiltä ja toinen isältä. Potilaallamme havaituista kahdesta c.35_36insA-mutaatiosta vain toinen löytyi hänen sisareltaan.
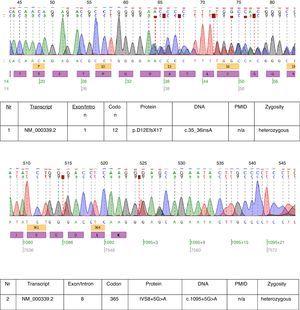
Kromatogrammit sairastuneen potilaan DNA:n sekvensoinnista.
Tältä potilaalta löydetyt mutaatiot miltei todistavat GS:n kliinisen diagnoosin. Frameshift-mutaation patogeneettinen merkitys on ilmeinen. Emme voi olla varmoja intronin 8 liitospaikkamutaation patogeneettisestä merkityksestä. Konsensusliitossekvenssi G (100 %), T (100 %), A (62 %), A (68 %), G (82 %), T (63 %) intronin 8 liitospaikan luovuttajan kohdalla ei täsmälleen vastaa konsensusta GTgAGc. Tällä potilaalla havaittu mutaatio poikkeaa sekvenssissä edelleen konsensuksesta (GtgAac). Myös in silico -analyysi mutation tasterin avulla pitää tätä mutaatiota tautia aiheuttavana.17
Keskustelu
Tubulopatiat ovat harvinaisia sairauksia. RenalTube-tietokannan18 mukaan yleisimpiä primaarisia tubulopatioita eurooppalaisessa väestössä ovat distaalinen munuaistubulaarinen asidoosi, Bartterin oireyhtymä, familiaalinen hypomagnesemia, johon liittyy hyperkalsiuria, ja GS.18 GS:n esiintyvyys on noin 25 tapausta miljoonaa ihmistä kohti.3 Koska GS on yksi yleisimmistä, monet nefrologit ja endokrinologit joutuvat luultavasti kohtaamaan GS-tapauksia uransa aikana. Potilaallamme oli tyypillinen GS:n kliininen oirekuva, ja hän reagoi hyvin hoitoon. GS:n ongelmana on, että liian vähäinen hypokalemia voi aiheuttaa kuoleman sydänpysähdyksen tai hengityslihasten halvaantumisen vuoksi. Vaikeaa neuromuskulaarista oireilua, kuten hypokalemiallista halvausta, esiintyy jopa 6 %:lla potilaista (yleisempää aasialaisilla potilailla).3
Perinnöllisillä tubulopatioilla on eräänlaiset ”peilikuvat” – diureettien aiheuttamat hankitut tubulopatiat. Kaikki diureetit (tarkemmin sanottuna: natriureetit, akvaretit tai glukureetit) aiheuttavat samanlaisia poikkeavuuksia kuin perinnöllisissä tubulopatioissa (taulukko 2). Lähes kaikissa tubulopatioissa esiintyy polyuriaa, ja kaliumpitoisuuden muutokset ovat yleisimpiä ongelmia. GS-potilailla esiintyy SLC12A3-geenin mutaatioita. Tämä geeni koodaa tiatsidille herkkää transporteria. Siksi GS muistuttaa kroonista tiatsidihoitoa.
Diureetit ja tubulopatiat.
| Yksittäinen perinnöllinen geenivirhe | Kaliumkonsentraatio | Oireet samankaltaisia kuin käyttö: | geenin | geenituotteen | geenituotteen sijainti | |
|---|---|---|---|---|---|---|
| Guibaud-Vainselin oireyhtymä (RTA 3) | ↓ | Asetatsoliamidi | CA2 | CA II | Proksimaalinen tubuli. | |
| Renaalinen glykosuria | – | SGLT2:n estäjätsa (Canagliflozin, Tofogliflotsiini, Dapagliflotsiini) | SLC5A2 | Natrium/glukoosi-kotransporter 2 (SGLT2) | Proksimaalinen tubulus | |
| Bartterin oireyhtymä | ↓ | Furosemidi | SLC12A1b | Na-K-2Cl-kotransporter (NKCC2) | Henlen silmukka | |
| Gitelmanin oireyhtymä | ↓ | Tiatsidi | SLC12A3 | Tiatsidi-herkkä Na-Cl-kotransporter | Distaalinen tubulus | |
| Diabetes insipidus | – | – | Vaptan | AVPR2, AQP2 | V2-reseptori | Keruukanava |
| Pseudohypoaldosteronizm, AD | ← | Spironolaktoni | NR3C2 | Spironolaktoni-sensitiivinen mineralokortikoidireseptori | Collecting ductus | |
| Pseudohypoaldosteronizm, AR | Amiloridi | SCNN1A, SCNN1B, SCNN1G | Amiloridi-sensitiivinen epiteelin natriumkanava | Collecting duct | ||
AR – autosomaalinen resessiivinen; AD – autosomaalinen dominantti.
SGLT2:n estäjät (glukureetit) ovat uusia lääkkeitä tyypin 2 diabeteksen hoidossa; hoidon aikana havaitaan osmoottiseen diureesiin liittyviä tapahtumia, kuten pollakiuriaa ja polyuriaa.
Vaikka Bartter-geenejä on erilaisia, tämä on se, joka jäljittelee furosemidiä.
Hypokalemia
GS tunnetaan myös nimellä familiaalinen hypokaleminen hypomagnesemia, koska hypokalemia on yleisin ilmiö. Potilaalle, jolla on hypokalemia ja epäillään tubulopatiaa, on varmistettava hyperkaliuria. Munuaisten kaliumhukka voidaan osoittaa laskemalla TKKG, FeK, satunnainen K/Cr-suhde tai 24 tunnin virtsanäytteenoton jälkeen (taulukko 1). Kaliurian arviointi olisi tehtävä, kun potilas ei käytä diureetteja eikä kaliumlisää ja kun kaliumpitoisuus on alhainen. Kun kaliumin erittyminen on alle 30 mmol/l ja TTKG on matala, hypokalemia johtuu ekstrarenaalisesta häviöstä tai transsellulaarisesta K+ -siirtymästä.
Kun hypokalemiaa sairastavan potilaan liiallinen kaliumin munuaishäviö varmistuu, jatkodiagnoosi perustuu pH:han ja verenpaineeseen. Monissa tapauksissa nämä yksinkertaiset tutkimukset riittävät diagnoosiin. Tärkeä ja yhä enemmän kiinnostusta herättävä ongelma on primaarinen aldosteronismi (PA). Joidenkin kliinisten tutkimusten mukaan PA on syynä yli 10 %:ssa arteriaalisesta hypertensiosta (AH), ja se on yleisempi potilailla, joilla AH on vastustuskykyinen verenpainelääkkeille.19 Hypokalemia on yksi PA:n ”klassisista” oireista, mutta se ei ole niin yleinen kuin luultiin.20 Koska AH:n diagnosoinnissa ei kuitenkaan ole olemassa kultaista standardia, RAA-akseli kannattaa muistaa potilailla, joilla on ateriapainetaudit (AH) ja hypokalemia.19
Hypomagnesemian diagnostiikka
Hypomagnesemia on yleinen poikkeavuus GS-potilailla, vaikka sitä ei havaita kaikissa tapauksissa.21 Normaalisti magnesemiaa sairastavilla GS-potilailla kliininen ilmentymä ja elektrolyyttipoikkeavuudet ovat lievempiä.22
Magnesiumionit suodattuvat kaliumionien tavoin vapaasti glomeruluksissa. Suodatetusta Mg2+:sta 10 % imeytyy proksimaaliseen tubulukseen, 50-70 % Henlen silmukan nousevaan haaraan. Distaalinen takaisinimeytyminen riippuu epiteelin Mg2+ TRPM6-kanavista. Magnesiuria riippuu oraalisesta saannista, ja FeMg olisi laskettava munuaisten Mg2+ -hukan toteamiseksi (FeMg=(virtsan magnesium × seerumin kreatiniini)/×100). FeMg alle 2 % viittaa huonoon saantiin, GI-hävikkiin tai Mg:n siirtymiseen soluihin. Yli 4 %:n FeMg viittaa munuaisten Mg2+ -hukkaan. Kun munuaisten Mg:n hukka on osoitettu, on laskettava satunnainen Ca/Cr-suhde. Virtsan Ca/Cr-suhde 0,3 on tyypillinen Bartterin oireyhtymässä esiintyvässä hypermagesuriassa, samoin kuin eristetyssä resessiivisessä hypomagnesemiassa, johon liittyy normokalsiuria, familiaalisessa hypomagnesemiassa, johon liittyy hyperkalsiuria ja nefrokalsinoosi, autosomaalisessa dominoivassa hypokalsemiassa, johon liittyy hyperkalsiuria, loop-diureettien hoidossa ja joidenkin nefrotoksiinien aiheuttamassa nefropatiassa.23
Magnesium- ja kaliumhomeostaasi ovat yhteydessä toisiinsa, eikä kaliumpuutosta voida korjata ennen hypomagnesaemian korjaamista.23 Keskimäärin potilaiden tulisi saada enintään 500mEq kaliumia, 4-5mg/kg/vrk 5-10mg magnesiumkloridia. Amiloridista (5-10mg/vrk) ja spironolaktonista (200-300mg) on apua.3
Kliiniset oireet ja laboratoriotulokset ovat olennaisia diagnoosin kannalta. Tapauksissa, joissa on tyypillinen fenotyyppi, jotkut kirjoittajat suosittelevat tiatsiditestin suorittamista diagnoosin varmistamiseksi.24 Esitetyssä tapauksessa geneettinen tutkimus paljasti kaksi uutta SLC12A3-geenin mutaatiota. Ne johtuvat todennäköisesti NCCT:n funktionaalisten mutaatioiden puuttumisesta.
Johtopäätökset
GS-oireyhtymä on yksi harvinaisista hypokalemian aiheuttajista, ja se näyttää olevan haaste lääkäreille. Osoitamme tässä artikkelissa, että jos muistaa hyvin yksinkertaisen lähestymistavan hypokalemiaan ja on tietoinen diureettien vaikutuksesta ja niiden samankaltaisuudesta perinnöllisten tubulopatioiden kanssa, diagnoosi voi olla varsin suoraviivainen. GS olisi erotettava muista tubulopatioista (sekä perinnöllisistä että hankituista) ja muista hypokalemian syistä (esim. Connin tauti). Sukuhistoria voi paljastaa oireettomia potilaita, joilla on GS. Sopiva hoito suojaa potilaita mahdollisesti vaarallisilta komplikaatioilta.