
Une brève introduction à ChIP-Seq

Les interactions protéine-ADN sont largement utilisées pour élucider les mécanismes qui sous-tendent la physiologie cellulaire. Le développement de la technologie du test d’immunoprécipitation de la chromatine (ChIP) a permis l’étude de tels mécanismes. Suite à d’autres développements, une alternative de séquençage profond (ChiP-Seq) est apparue, qui offre des avantages en termes de spécificité et de sensibilité.
Une expérience ChIP-Seq commence par une réticulation de la cellule entière avec du formaldéhyde, suivie d’une sonication et d’une isolation de l’ADN. Ensuite, on procède à l’immunoprécipitation du complexe ADN-protéine, qui consiste en la liaison d’anticorps à des protéines spécifiques. Les immunocomplexes formés sont précipités et purifiés. Enfin, l’ADN est séquencé, ce qui génère des données à haute résolution sur les sites enrichis. Cette approche, associée à un pipeline ChIP-seq bien établi, permet aux chercheurs de capturer les facteurs de transcription de l’ADN, les sites de modification des histones, les altérations épigénétiques et les signatures des réseaux de régulation des gènes.
Pertinence clinique et applications
Les déséquilibres épigénétiques à travers les maladies et les conditions de santé peuvent impliquer une modification des histones et des facteurs de transcription altérés. Dans ce contexte, les études ChIP-Seq ont été utilisées pour élucider les mécanismes moléculaires pathologiques qui sous-tendent le cancer et d’autres maladies. L’analyse ChIP-seq contribue également à la compréhension du rôle des facteurs de transcription au cours des maladies. En effet, certains transcrits semblent être altérés au cours des manifestations du phénotype clinique.
Vue d’ensemble du pipeline ChIP-Seq
Le pipeline d’analyse ChIP-Seq est le composant principal des projets d’interaction ADN-protéine et se compose de plusieurs étapes, notamment le traitement des données brutes, l’analyse de contrôle de la qualité, l’alignement sur le génome de référence, le contrôle de la qualité des lectures alignées, l’appel de pic, l’annotation et la visualisation. Cependant, il est essentiel de disposer d’un plan expérimental bien pensé pour obtenir des résultats de haute qualité lors d’une expérience ChIP-seq. Avant de commencer votre analyse, il est essentiel de prendre en compte des paramètres tels que les réplicats d’échantillons, les groupes de contrôle, les kits de séquençage et les plateformes de séquençage.
Contrôle de qualité
Tous les rapports Basepair fournissent des scores de qualité pour aider à découvrir les problèmes potentiels de séquençage ou de contamination dans vos données d’entrée.
L’étape de contrôle de qualité (CQ) vise à évaluer la qualité des données à haut débit générées par le séquençage. Cette étape est similaire à celles réalisées dans les analyses ADN-seq et ARN-seq. Ici, les principaux paramètres évalués sont la qualité des séquences et des bases, le contenu en GC, la présence d’adaptateurs de séquençage et les séquences surreprésentées. L’un des programmes les plus couramment utilisés pour ce type d’analyse est FastQC. De plus, si des séquences de faible qualité sont identifiées, elles peuvent être supprimées ultérieurement lors de l’étape d’élagage. Bien qu’il s’agisse d’une étape facultative, le détourage améliore la qualité des données en ne conservant que les lectures de haute qualité.
Alignement
Après la mesure du QC, les lectures ChIP-Seq sont alignées sur un génome de référence. La cartographie des lectures permet aux chercheurs d’identifier l’origine d’une séquence de lecture dans le génome. Les outils logiciels d’alignement populaires utilisés comprennent Bowtie et BWA, qui sont tous deux utilisés dans les pipelines ChIP-seq de Basepair. Les deux outils cartographient les séquences à faible divergence par rapport à un génome de référence.
Le flux de comptage des lectures aide à fournir une vue d’ensemble des lectures utilisables à la fin des processus de rognage, d’alignement et de déduplication. Pensez à la figure comme à une chaîne de montage d’analyse de données : entrée de données brutes, obtention d’une sortie de lectures utilisables.
Contrôle de qualité des lectures alignées
L’étape suivante consiste en une inférence QC de l’ensemble de données alignées. Au cours du processus de cartographie, les doublons de lecture introduits par l’amplification PCR et le séquençage provoquent des biais lors de l’appel de pic et de l’analyse d’enrichissement. Basepair utilise l’outil Picard pour supprimer les doublons. Une fois les doublons supprimés, vous devez évaluer la fraction non redondante (NRF) des lectures alignées. La NRF mesure les lectures uniques qui correspondent au génome de référence. Les expériences ChIP-seq idéales devraient avoir moins de trois lectures par position.
Peak Calling
L’étape de peak calling détecte les régions d’interaction protéine-ADN enrichies sur le génome. Le pipeline ChIP-seq de Basepair utilise MACS2 pour effectuer cette analyse. Dans MACS2, l’appel de pic est effectué sur la base de trois étapes principales : l’estimation des fragments, suivie de l’identification des paramètres de bruit locaux, puis de l’identification des pics. En sortie de cette étape, les utilisateurs obtiennent un tableau final contenant des informations sur les pics, telles que le score d’enrichissement, la valeur -log10p, la valeur -log10q et la position par rapport au début du pic. L’utilisation d’échantillons de contrôle est fortement recommandée dans cette étape pour la comparaison avec l’ensemble de données cible étudié. Gardez à l’esprit que de bons groupes de contrôle apportent des résultats plus fiables.
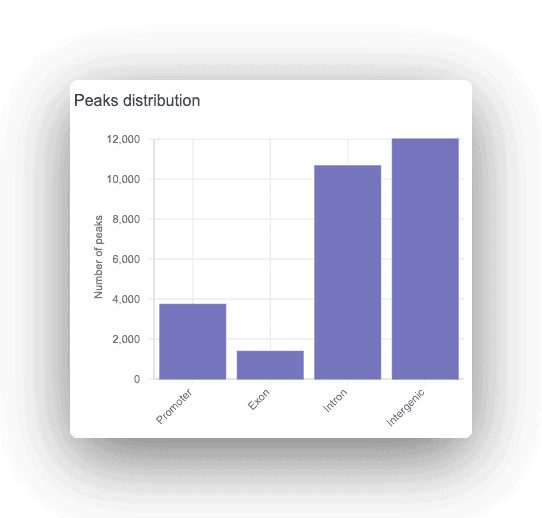
Chaque pic est annoté comme promoteur, intronique ou intergénique, avec le gène correspondant affiché. Pour tous les pics trouvés, une analyse des motifs est effectuée pour trouver les sites de liaison des facteurs de transcription surreprésentés.
Vue d’ensemble des résultats
Un pipeline ChIP-seq peut fournir non seulement des informations sur l’état de la chromatine mais aussi sur la liaison des facteurs de transcription dans un contexte de gènes ou de loci déterminé. L’occurrence des modifications des histones et des facteurs de transcription dans les régions régulatrices de l’ADN peut constituer une signature épigénétique spécifique à une condition. Ainsi, les perturbations épigénétiques peuvent être associées à des phénotypes cliniques. Par exemple, l’hétérogénéité des états de la chromatine peut entraîner une résistance au traitement dans le cancer du sein. Ces cellules ont tendance à perdre les marqueurs de modifications répressives des histones et à augmenter davantage l’expression des gènes connus pour favoriser la résistance au traitement du cancer.
Analyse des pics, des motifs et des voies dans le pipeline d’analyse ChIP-Seq
L’identification de l’enrichissement des facteurs de transcription en motifs est utilisée pour élucider si les facteurs de transcription coopèrent ou sont en compétition dans une région donnée. L’identification des pics dans les régions de motifs d’ADN peut améliorer l’interprétation des résultats expérimentaux. Ensemble, les analyses des pics et des motifs donnent un aperçu de ce qui peut se produire dans une cellule. L’intégration des enrichissements des pics et des motifs permet d’obtenir un paysage épigénomique avec des conséquences biologiques possibles. En outre, l’analyse des voies est utilisée pour identifier les protéines dans une voie. Les investigations et les conclusions sont formulées en fonction de la présence d’une protéine.
Visualisation des données
Les données résultantes d’un pipeline ChIP-seq peuvent être visualisées en utilisant un navigateur de génome. Les rapports Basepair incluent un navigateur de génome IGV2 intégré qui vous permet d’interagir avec vos données. Les données peuvent également être visualisées à l’aide de cartes thermiques, qui sont des infographies d’intensité représentative basées sur la densité des données et qui montrent la présence ou l’absence de marques spécifiques. D’autres graphiques utilisés ici comprennent le tracé d’enrichissement, l’upSet et le tracé de couverture, qui calcule et affiche la couverture des régions de pics sur le génome.
Le navigateur de génome est un excellent outil pour visualiser vos données génomiques brutes. Il est intégré dans chaque rapport d’analyse ChIP-seq sur Basepair.
1. Grosselin, K., A. Durand, et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet, v.51, n.6, Jun, p.1060-1066. 2019.
2. Northrup, D. L. e K. Zhao. Application de ChIP-Seq et de techniques connexes à l’étude de la fonction immunitaire. Immunité, v.34, n.6, 24 juin, p.830-42. 2011.
3. Park, S. J., J. H. Kim, et al. A ChIP-Seq Data Analysis Pipeline Based on Bioconductor Packages. Genomics Inform, v.15, n.1, Mar, p.11-18. 2017.
4. Pepke, S., B. Wold, et al. Calcul pour les études ChIP-seq et RNA-seq. Nat Methods, v.6, n.11 Suppl, Nov, p.S22-32. 2009.
5. Satoh, J., N. Kawana, et al. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul Syst Bio, v.7, p.139-52. 2013.