
Introduction
L’hypokaliémie est un problème clinique courant dans la pratique des endocrinologues et des néphrologues. Il existe de nombreuses causes évidentes d’hypokaliémie telles que la diarrhée, les vomissements ou l’abus de diurétiques. D’autres causes, comme les tubulopathies, sont rarement observées et leur diagnostic est plus difficile. Il existe de nombreuses tubulopathies héréditaires et acquises qui provoquent une hypokaliémie, parfois grave et potentiellement mortelle.1
Une cause relativement courante mais négligée d’hypokaliémie est le syndrome de Gitelman (SG).2 Il s’agit d’une tubulopathie récessive avec perte de sel causée par la mutation du gène SLC12A3. Le gène SLC12A3 code pour le transporteur sensible aux thiazides NCCT (co-transporteur du chlorure de sodium). Le NCCT est situé dans les cellules tubulaires convoluées distales (DCC), qui sont responsables de 7 à 10 % de l’absorption tubulaire d’électrolytes.3
Les anomalies de laboratoire les plus graves trouvées dans le GS sont l’hypokaliémie et l’hypomagnésémie causées par une déperdition rénale de K+ et de Mg2+. D’autres changements typiques sont l’alcalose métabolique, l’hypocalciurie et l’hyperaldostéronisme hyperrénémique.4 Une hypophosphatémie légère à modérée est fréquemment observée.5 Une hypophosphatémie sévère avec une hyponatrémie sévère a également été rapportée.6,7
Les premiers symptômes du SG apparaissent chez les enfants ou les jeunes adultes ayant une croissance normale et des antécédents de comportements de dévoration du sel (enfants désireux de consommer des cornichons ou de la saumure, des concombres, des oranges et des citrons salés, enfants léchant le sel des chips, etc.).8 La présentation clinique varie selon les patients. Certains sont asymptomatiques, mais d’autres développent des complications potentiellement mortelles. Les hommes présentent un phénotype plus sévère que les femmes.8 Les symptômes les plus courants sont les crampes et la faiblesse musculaires, la constipation, la nycturie, la polyurie, la soif, la polydipsie, les arythmies cardiaques, les paresthésies et un appétit accru pour le sel. L’hypotension artérielle est fréquente et, dans de nombreux cas, constitue le symptôme le plus marquant, mais une hypertension peut survenir dans la population vieillissante du GS.8 La corrélation entre les anomalies biochimiques et les symptômes n’est pas forte.9 Le GS n’interfère pas avec l’humeur et les relations sociales des enfants.9,10 Sinon, les symptômes sont plus fréquents chez les adultes et peuvent avoir un impact négatif sur leur qualité de vie. Quarante-cinq pour cent des patients atteints du GS considèrent leurs symptômes comme un problème modéré à important.11 L’épuisement extrême, la faiblesse musculaire, les paresthésies, la fatigue sévère et l’hypotension sont associés à une réduction légère à sévère des activités quotidiennes.9
La prévalence estimée du GS est de 1:40 0008 et la prévalence de l’hétérozygotie est d’au moins 1% dans la population européenne. Plus de 180 mutations différentes dans SLC12A3 ont été décrites jusqu’à présent.12
Rapport de cas
Un homme de 26 ans a été admis à l’hôpital en raison d’une incidence de syncope, de faiblesse généralisée et musculaire et de crampes musculaires. Les antécédents du patient ont révélé un épisode de syncope avec un taux de potassium de 3,16mmol/l. Lors d’un suivi ultérieur en consultation externe, on a observé une incidence récurrente d’hypokaliémie (la valeur la plus basse étant de 2,6mmol/l). La pression artérielle était normale 110/80, la fréquence cardiaque était de 72 par minute ; il n’y avait pas de changement dans l’examen physique. Pas de résultats neurologiques, poids 74kg, taille 178cm. A l’admission à l’hôpital, l’évaluation de laboratoire a montré une légère hypokaliémie (K+ 3.0mmol/l), une hypomagnésémie (Mg2+ 1.36mg/dl), une hypocalciurie (
mg/24h), et une alcalose métabolique (HCO3- 29.7mmol/l, BE 5.3mmol/l). La fonction rénale était bonne avec un DFGe>60ml/min. Les examens d’imagerie étaient sans particularité, de même que l’ECG.
Des examens complémentaires ont confirmé que l’hypokaliémie était due à une déperdition rénale de potassium. La déperdition potassique sur 24 heures, le gradient transtubulaire de K+ (TTKG), l’excrétion fractionnelle de K+ (FeK) et le rapport K/créatinine aléatoire (K/Cr) étaient typiques de l’hyperkaliurie et sont présentés dans le tableau 1.13-15 La magnésiurie quotidienne était de 64mg/24h, avec une excrétion fractionnelle de magnésium (FeMg) de 15%.
Les valeurs diagnostiques d’une hypokaliémie.13-15
| Valeurs diagnostiques d’une kaliurèse inappropriée | Patient | |
|---|---|---|
| K+ urinaire | >20mmol/24h Si le K+ sérique est de 3.5mmol/l |
57mmol/l (K+ sérique=3.0mmol/l) |
| FeK=×100% | >6,5% Si le sérum K+3.5mmol/l |
12% (sérum K+=3.0mmol/l) |
| TTKG= | TTKG>2 Si le sérum K+3.5mmol/l |
7,3 (sérum K+=3,0mmol/l) |
| uK/Cr – rapport aléatoire K+ urinaire/créatinine | uK/Cr>15mmol/g Si sérum K+3.5mmol/l |
67mmol/g (K+ sérique=3.0mmol/l) |
Bien que la pression artérielle ait été normale, des tests hormonaux pour exclure la maladie de Conn et le CT abdominal ont été effectués, et n’ont pas révélé d’anomalies dans les glandes surrénales. Un hyperaldostéronisme secondaire avec des niveaux d’aldostérone 289pg/ml (plage normale 20-180) et de rénine 205mIU/ml (plage normale 2,8-39,9) étaient typiques du GS.
Le patient a été diagnostiqué comme GS sur la base d’un phénotype clinique. Après le diagnostic, un traitement avec des suppléments de potassium et de magnésium a été introduit. Il a reçu 20mmol de chlorure de potassium et 18mmol de pyrrolidone carboxylate de magnésium, avec une amélioration clinique. L’histoire familiale a révélé une légère hypokaliémie asymptomatique chez la sœur du patient, âgée de 35 ans. Mais les examens n’ont montré ni hyperkaliurie ni hypomagnésémie. Le GS n’a pas été diagnostiqué dans ce cas.
Analyse génétique
Selon l’analyse génétique, algorithme proposé par Nozu16 Le gèneSLC12A3 devrait être testé chez les patients présentant une alcalose métabolique hypokaliémique, avec une naissance à terme, un poids normal sans néphrocalcinose, avec une hypocalcurie et une hypomagnésémie.
Dans le cas décrit, un test génétique a été effectué et deux mutations hétérozygotes : c.35_36insA et c.1095+5G>A ont été trouvées dans le transcrit NM_000339.2 du gène SLC12A3 (Fig. 1). Les deux mutations ne figurent pas encore dans la version disponible de la base de données HGMD(r) . La première mutation a également été trouvée chez la mère du patient et la seconde chez le père. Une seule des deux mutations identifiées chez notre patient c.35_36insA a été retrouvée chez sa sœur.
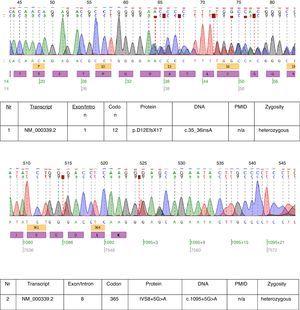
Chromatogrammes du séquençage de l’ADN du patient affecté.
Les mutations trouvées chez ce patient prouvent presque le diagnostic clinique de GS. La pertinence pathogénétique de la mutation de décalage de cadre est évidente. Nous ne pouvons pas être sûrs de la pertinence pathogénétique de la mutation du site d’épissage de l’intron 8. La séquence d’épissage consensus G (100%), T (100%), A (62%), A (68%), G (82%), T (63%) au niveau du donneur d’épissage de l’intron 8 ne correspond pas exactement au consensus GTgAGc. La mutation trouvée chez ce patient dévie davantage la séquence du consensus (GtgAac). De plus, l’analyse in silico effectuée par le testeur de mutation considère que cette mutation est à l’origine de la maladie.17
Discussion
Les tubulopathies sont des maladies rares. Selon la base de données RenalTube18, les tubulopathies primaires les plus courantes dans la population européenne sont l’acidose tubulaire rénale distale, le syndrome de Bartter, l’hypomagnésémie familiale avec hypercalciurie et le GS.18 La prévalence du GS est d’environ 25 cas pour 1 million d’habitants.3 Le GS étant l’une des plus courantes, il est probable que de nombreux néphrologues et endocrinologues seront confrontés à des cas de GS au cours de leur carrière. Notre patient avait une présentation clinique typique du GS et a bien répondu au traitement. Le problème du GS est qu’une hypokaliémie négligée peut entraîner la mort par arrêt cardiaque ou paralysie des muscles respiratoires. Le symptôme neuromusculaire grave tel que la paralysie hypokaliémique se produit chez jusqu’à 6% des patients (plus fréquent chez les patients asiatiques).3
Les tubulopathies héréditaires ont une sorte de « miroir » – les tubulopathies acquises causées par les diurétiques. Tous les diurétiques (pour être précis : natriurétiques, acquarétiques ou glucurétiques) provoquent des anomalies similaires à celles trouvées dans les tubulopathies héréditaires (Tableau 2). Presque dans chaque tubulopathie, une polyurie se produit, et les modifications du taux de potassium sont parmi les problèmes les plus courants. Chez les patients atteints de GS, on trouve des mutations du gène SLC12A3. Ce gène code pour le transporteur sensible aux thiazides. Le GS ressemble donc à un traitement chronique par les thiazides.
Diurétiques et tubulopathies.
| Défaut génétique unique héréditaire | Concentration de potassium | Symptômes similaires à l’utilisation de : | Gène | Produit génique | Localisation du produit génique |
|---|---|---|---|---|---|
| Syndrome de Guibaud-…Vainsel (RTA 3) | ↓ | Acetazolamide | CA2 | CA II | Tubule proximal |
| Glycosurie rénale | – | Inhibiteurs du SGLT2a (Canagliflozine, Tofogliflozine, Dapagliflozine) | SLC5A2 | Cotransporteur 2 du sodium/glucose (SGLT2) | Tubule proximal |
| Syndrome de Bartter | ↓ | Furosémide | SLC12A1b | Na-…K-2Cl (NKCC2) | Boucle de Henle |
| Syndrome de Gitelman | ↓ | Tiazide | SLC12A3 | Tiazide-.cotransporteur Na-Cl sensible au thiazide | Tubule distal |
| Diabète insipide | – | Vaptan | AVPR2, AQP2 | Récepteur V2 | Canal collecteur |
| Pseudohypoaldostéronisme, AD | ← | Spironolactone | NR3C2 | Récepteur minéralocorticoïde sensible à la spironolactone | Canal collecteur |
| Pseudohypoaldosteronizm, AR | Amiloride | SCNN1A, SCNN1B, SCNN1G | Canal sodique épithélial sensible à l’amiloride | Canal collecteur | |
AR – autosomique récessif ; AD – autosomique dominant.
Les inhibiteurs du SGLT2 (glucurétiques) sont les nouveaux médicaments pour la prise en charge du diabète de type 2 ; pendant le traitement, on observe des événements liés à la diurèse osmotique tels que la pollakiurie et la polyurie.
Bien qu’il existe différents gènes de Bartter, celui-ci est celui qui mime le furosémide.
Approche de l’hypokaliémie
La SG est aussi appelée hypomagnésémie hypokaliémique familiale, car l’hypokaliémie est le phénomène le plus fréquent. Chez un patient présentant une hypokaliémie et une suspicion de tubulopathie, il faut confirmer l’hyperkaliurie. La perte rénale de potassium peut être prouvée en calculant le TKKG, le FeK, le rapport aléatoire K/Cr ou après une collecte d’urine sur 24 heures (tableau 1). L’évaluation de la kaliurie doit être effectuée lorsque le patient ne prend ni diurétique ni supplémentation en potassium et lorsque le taux de potassium est bas. Lorsque l’excrétion de potassium est inférieure à 30mmol/l et que le TTKG est faible, l’hypokaliémie est due à une perte extrarénale ou à un déplacement transcellulaire de K+.
Lorsque la perte rénale excessive de potassium chez un patient présentant une hypokaliémie est confirmée, le diagnostic ultérieur repose sur le pH et la pression artérielle. Dans de nombreux cas, ces études simples seront suffisantes pour le diagnostic. Un problème important qui suscite un intérêt croissant est l’aldostéronisme primaire (AP). Certaines études cliniques ont suggéré que l’AP est la cause de plus de 10% de l’hypertension artérielle (AH), et qu’il est plus fréquent chez les patients dont l’AH est résistante aux agents antihypertenseurs.19 L’hypokaliémie est l’un des symptômes « classiques » de l’AP, mais elle n’est pas aussi fréquente qu’on le pensait.20 Pourtant, comme il n’y a pas d’étalon-or dans le diagnostic de l’AP, il est utile de se souvenir de l’axe RAA chez les patients atteints d’AH et d’hypokaliémie.19
Diagnostic de l’hypomagnésémie
L’hypomagnésémie est une anomalie courante chez les patients atteints de GS, bien qu’elle ne soit pas observée dans tous les cas.21 Chez les patients normomagnésémiques atteints de GS, la manifestation clinique et les anomalies électrolytiques sont plus légères.22
Similaires aux ions potassium, les ions magnésium sont librement filtrés par les glomérules. 10% du Mg2+ filtré est absorbé par le tubule proximal, 50-70% dans le membre ascendant de l’anse de Henle. La réabsorption distale dépend des canaux épithéliaux Mg2+ TRPM6. La magnésiurie dépend de l’apport oral et le FeMg doit être calculé pour établir la perte rénale de Mg2+ (FeMg=(magnésium urinaire×créatinine sérique)/×100). Un FeMg inférieur à 2 % suggère un apport insuffisant, des pertes gastro-intestinales ou un transfert de Mg dans les cellules. Un FeMg supérieur à 4 % suggère une perte rénale de Mg2+. Lorsque la perte rénale de Mg est prouvée, le rapport Ca/Cr aléatoire doit être calculé. Le rapport Ca/Cr urinaire de 0,3 est typique de l’hypermagesurie dans le syndrome de Bartter, de même que l’hypomagnésémie récessive isolée avec normocalciurie, l’hypomagnésémie familiale avec hypercalciurie et néphrocalcinose, l’hypocalcémie autosomique dominante avec hypercalciurie, le traitement par diurétiques de l’anse et la néphropathie causée par certaines néphrotoxines23.
L’homéostasie du magnésium et du potassium sont liées l’une à l’autre, et la déplétion potassique ne peut être corrigée avant la correction de l’hypomagnésémie.23 En moyenne, les patients doivent recevoir jusqu’à 500mEq de potassium, 4-5mg/kg/jour de 5-10mg de chlorure de magnésium. L’amiloride (5-10mg/jour) et le spironolacton (200-300mg) sont utiles.3
Les symptômes cliniques et les résultats de laboratoire sont essentiels pour le diagnostic. Dans les cas présentant un phénotype typique, certains auteurs recommandent d’effectuer un test aux thiazides pour confirmer le diagnostic.24 Dans le cas présenté, l’enquête génétique a révélé deux nouvelles mutations du gène SLC12A3. Elles sont probablement causées par l’absence de mutations fonctionnelles du NCCT.
Conclusions
Le syndrome SG est l’une des rares causes d’hypokaliémie, et il semble être un défi pour les médecins. Nous montrons dans cet article que si l’on se souvient d’une approche très simple de l’hypokaliémie et que l’on connaît l’action des diurétiques et leur similitude avec les tubulopathies héréditaires, le diagnostic pourrait être assez simple. Le GS doit être différencié des autres tubulopathies (héréditaires ou acquises) et des autres causes d’hypokaliémie (par exemple, la maladie de Conn). Les antécédents familiaux peuvent révéler des patients asymptomatiques atteints du GS. Un traitement approprié protège les patients de complications potentiellement dangereuses.