
En kort introduktion till ChIP-Seq

Protein-DNA-interaktioner används i stor utsträckning för att belysa de mekanismer som ligger till grund för cellfysiologi. Utvecklingen av tekniken för kromatinimmunutfällning (ChIP) gjorde det möjligt att studera sådana mekanismer. Efter ytterligare utveckling uppstod ett alternativ för djup sekvensering (ChiP-Seq) som erbjuder fördelar när det gäller specificitet och känslighet.
Ett ChIP-Seq-experiment börjar med en tvärbindning av hela celler med formaldehyd, följt av sonikation och DNA-isolering. Därefter utförs immunutfällning av DNA-proteinkomplexet, som består av antikroppar som binder till specifika proteiner. De immunokomplex som bildas fälls ut och renas. Slutligen sekvenseras DNA, vilket genererar högupplösta data om berikade platser. Detta tillvägagångssätt, tillsammans med en väletablerad ChIP-seq-pipeline, gör det möjligt för forskare att fånga DNA-transkriptionsfaktorer, histonmodifieringsställen, epigenetiska förändringar och signaturer för genetiska regleringsnätverk.
Klinisk relevans och tillämpningar
Epigenetiska obalanser i olika sjukdoms- och hälsotillstånd kan inbegripa histonmodifiering och förändrade transkriptionsfaktorer. Här har ChIP-Seq-studier använts för att belysa patologiska molekylära mekanismer som ligger till grund för cancer och andra sjukdomar. ChIP-seq-analyser bidrar också till förståelsen av transkriptionsfaktorernas roll under sjukdomar. Faktum är att vissa transkript tycks förändras under kliniska fenotypmanifestationer.
Översikt över ChIP-Seq-pipeline
Pipeline för ChIP-Seq-analys är huvudkomponenten i DNA-proteininteraktionsprojekt och består av flera steg, bland annat bearbetning av rådata, kvalitetskontrollanalys, anpassning till referensgenomet, kvalitetskontroll av de anpassade läsningarna, peak calling, annotering och visualisering. Att ha en genomtänkt experimentell utformning är dock avgörande för att få högkvalitativa resultat i ett ChIP-seq-experiment. Innan du påbörjar din analys är det viktigt att överväga parametrar som provreplikat, kontrollgrupper, sekvenseringskit och sekvenseringsplattformar.
Kvalitetskontroll
Alla Basepair-rapporter ger kvalitetspoäng för att hjälpa till att avslöja potentiella sekvenseringsproblem eller kontaminering i dina indata.
Steget för kvalitetskontroll (QC) syftar till att utvärdera kvaliteten på data med hög genomströmning som genereras från sekvensering. Detta steg liknar de steg som utförs i DNA-seq- och RNA-seq-analyser. Här omfattar de viktigaste mätvärdena som utvärderas sekvens- och baskvalitet, GC-innehåll, närvaro av sekvenseringsadaptrar och överrepresenterade sekvenser. Ett av de vanligaste programmen för denna typ av analys är FastQC. Om sekvenser av låg kvalitet identifieras kan de dessutom senare tas bort under trimmningssteget. Även om det är ett valfritt steg förbättrar trimning datakvaliteten genom att endast högkvalitativa reads behålls.
Alignment
Efter QC-mätning anpassas ChIP-Seq-reads till ett referensgenom. Med hjälp av läskartläggning kan forskarna identifiera ursprunget för en lässekvens i genomet. Populära programvaruverktyg för justering som används är Bowtie och BWA, som båda används i Basepairs ChIP-seq-pipelines. Båda verktygen kartlägger lågt divergerande sekvenser mot ett referensgenom.
Flödet för läsuppräkning hjälper till att ge en helhetsbild av användbara läsuppräkningar i slutet av processerna för trimning, justering och deduplicering. Tänk på figuren som ett löpande band för dataanalys: inmatning av rådata, erhåller ett resultat av användbara läsningar.
Kvalitetskontroll av de anpassade läsningarna
Nästa steg består av QC-inferens av den anpassade datamängden. Under kartläggningsprocessen orsakar läsdubbletter som introduceras av PCR-amplifiering och sekvensering bias under peak calling och anrikningsanalysen. Basepair använder Picard-verktyget för att ta bort dubbletter. När duplikat har tagits bort bör du utvärdera Non-Redundant Fraction (NRF) av de anpassade läsningarna. NRF mäter de unika läsningar som mappas till referensgenomet. Ideala ChIP-seq-experiment bör ha mindre än tre läsningar per position.
Peak Calling
Steget Peak Calling upptäcker berikade protein-DNA-interaktionsregioner på genomet. Basepairs ChIP-seq-pipeline använder MACS2 för att utföra denna analys. I MACS2 utförs peak calling baserat på tre huvudsteg: fragmentbedömning, följt av identifiering av lokala brusparametrar och därefter identifiering av toppar. Som resultat av detta steg får användarna en slutlig tabell med information om toppar, t.ex. anrikningsvärde, -log10p-värde, -log10q-värde och positionen till toppens start. Användning av kontrollprover rekommenderas starkt i detta steg för jämförelse med det undersökta måldatasetet. Tänk på att bra kontrollgrupper ger mer tillförlitliga resultat.
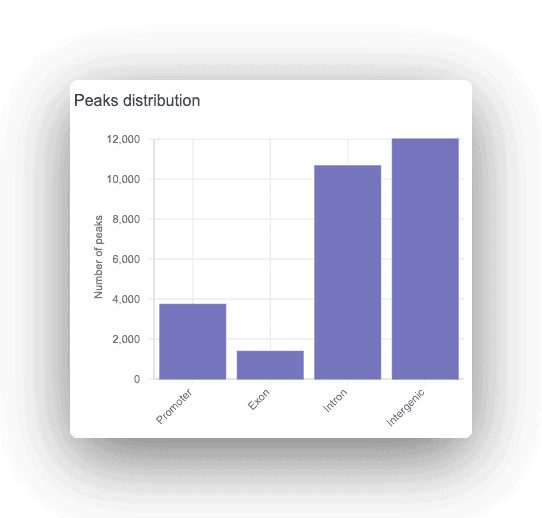
Varje topp är annoterad som promotor, intronisk eller intergenisk, och motsvarande gen visas. För alla toppar som hittas görs en motivanalys för att hitta överrepresenterade bindningsställen för transkriptionsfaktorer.
Översikt över resultat
En ChIP-seq-pipeline kan ge inte bara information om kromatintillståndet utan också om bindning av transkriptionsfaktorer i en bestämd gen eller loci-kontext. Förekomsten av histonmodifieringar och transkriptionsfaktorer i DNA-regulatoriska regioner kan utgöra en tillståndsspecifik epigenetisk signatur. Epigenetiska störningar kan således förknippas med kliniska fenotyper. Till exempel kan heterogenitet i kromatintillstånd leda till behandlingsresistens vid bröstcancer. Dessa celler tenderar att förlora repressiva histonmodifieringsmarkörer och ytterligare öka uttrycket av gener som är kända för att främja resistens mot cancerbehandling.
Peak, Motif and Pathway Analysis in ChIP-Seq Analysis Pipeline
Identifieringen av motivtranskriptionsfaktorsanrikning används för att klarlägga om transkriptionsfaktorer samarbetar eller konkurrerar i en viss region. Identifieringen av toppar i DNA-motivregioner kan förbättra tolkningen av de experimentella resultaten. Tillsammans ger både topp- och motivanalyser insikt i vad som kan hända i en cell. Integrationen av toppar och motivberikningar resulterar i ett epigenomiskt landskap med möjliga biologiska konsekvenser. Vidare används väganalyser för att identifiera proteiner i en väg. Undersökningar och slutsatser formuleras utifrån en proteinnärvaro.
Datavisualisering
Resultatdata från en ChIP-seq-pipeline kan visualiseras med hjälp av en genombrowser. Basepair-rapporter innehåller en inbäddad IGV2-genombrowser som gör att du kan interagera med dina data. Data kan alternativt visualiseras med hjälp av heatmaps, som är representativa intensitetsinfografiker baserade på datatäthet som visar närvaron eller frånvaron av specifika markeringar. Andra grafiker som används här är anrikningsplot, upSet och coverage plot, som både beräknar och visar täckningen av toppregioner över genomet.
Genombrowsern är ett utmärkt verktyg för att visualisera dina rågenomiska data. Den är inbyggd i varje ChIP-seq-analysrapport på Basepair.
1. Grosselin, K., A. Durand, et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet, v.51, n.6, Jun, s.1060-1066. 2019.
2. Northrup, D. L. e K. Zhao. Tillämpning av ChIP-Seq och relaterade tekniker för studier av immunfunktion. Immunity, v.34, n.6, jun 24, s.830-42. 2011.
3. Park, S. J., J. H. Kim, et al. A ChIP-Seq Data Analysis Pipeline Based on Bioconductor Packages. Genomics Inform, v.15, n.1, mars, s.11-18. 2017.
4. Pepke, S., B. Wold, et al. Computation for ChIP-seq and RNA-seq studies. Nat Methods, v.6, n.11 Suppl, Nov, s.S22-32. 2009.
5. Satoh, J., N. Kawana, et al. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul Syst Bio, v.7, s.139-52. 2013.