
Uma breve introdução ao ChIP-Seq

Interações proteína-DNA são amplamente utilizadas para elucidar os mecanismos subjacentes à fisiologia celular. O desenvolvimento da tecnologia do ensaio de imunoprecipitação de cromatina (ChIP) permitiu o estudo de tais mecanismos. Após um maior desenvolvimento, surgiu uma alternativa de sequenciamento profundo (ChiP-Seq), que oferece vantagens em termos de especificidade e sensibilidade.
A experiência ChIP-Seq começa com uma ligação cruzada de células inteiras com formaldeído, seguida de sonicação e isolamento de DNA. Depois disso, é realizada a imunoprecipitação do complexo DNA-proteína, que consiste na ligação de anticorpos a proteínas específicas. Os imunocomplexos formados são precipitados e purificados. Finalmente, o ADN é sequenciado, gerando dados de alta resolução de locais enriquecidos. Esta abordagem, juntamente com um bem estabelecido gasoduto ChIP-seq, permite aos pesquisadores capturar fatores de transcrição de DNA, locais de modificação de histone, alterações epigenéticas e assinaturas de redes reguladoras de genes.
Pertinência e Aplicações Clínicas
Desequilíbrios epigenéticos entre doenças e condições de saúde podem envolver modificação de histone e fatores de transcrição alterados. Aqui, estudos ChIP-Seq têm sido usados para elucidar mecanismos moleculares patológicos subjacentes ao câncer e outras doenças. A análise ChIP-Seq também contribui para a compreensão do papel dos fatores de transcrição durante as doenças. De fato, algumas transcrições parecem ser alteradas durante manifestações fenotípicas clínicas.
Visão geral do pipeline ChIP-Seq
O pipeline de análise ChIP-Seq é o principal componente dos projetos de interação DNA-proteína e consiste em várias etapas, incluindo processamento de dados brutos, análise de controle de qualidade, alinhamento ao genoma de referência, verificação de qualidade das leituras alinhadas, chamada de pico, anotação e visualização. No entanto, ter um projeto experimental bem pensado é crucial para obter resultados de alta qualidade em um experimento ChIP-seq. Antes de iniciar sua análise, é essencial considerar parâmetros como réplicas de amostras, grupos de controle, kits de sequenciamento e plataformas de sequenciamento.
Controle de Qualidade
Todos os relatórios Basepair fornecem escores de qualidade para ajudar a descobrir possíveis problemas de sequenciamento ou contaminação nos seus dados de entrada.
O passo Controle de Qualidade (CQ) tem como objetivo avaliar a qualidade dos dados de alto rendimento gerados pelo sequenciamento. Esta etapa é semelhante àquelas realizadas nas análises de DNA-seq e RNA-seq. Aqui, as principais métricas avaliadas incluem sequência e qualidade de base, conteúdo GC, presença de adaptadores de sequenciamento e sequências sobre-representadas. Um dos programas mais utilizados para este tipo de análise é o FastQC. Além disso, se sequências de baixa qualidade forem identificadas, elas podem ser removidas posteriormente durante a etapa de corte. Embora seja um passo opcional, o corte de linha melhora a qualidade dos dados ao reter somente leituras de alta qualidade.
Alinhamento
Após a medição do CQ, as leituras ChIP-Seq são alinhadas a um genoma de referência. O mapeamento de leitura permite aos pesquisadores identificar a origem de uma sequência de leitura no genoma. As ferramentas populares de software de alinhamento utilizadas incluem Bowtie e BWA, ambos utilizados nos pipelines ChIP-seq da Basepair. Ambas as ferramentas mapeiam seqüências de baixa divergência contra um genoma de referência.
O fluxo de contagem de leitura ajuda a fornecer uma visão geral das leituras utilizáveis no final dos processos de corte, alinhamento e deduplicação. Pense na figura como uma linha de montagem de análise de dados: insira dados brutos, obtenha uma saída de leituras utilizáveis.
Verificação da qualidade das leituras alinhadas
O próximo passo consiste na inferência de QC do conjunto de dados alinhados. Durante o processo de mapeamento, as leituras duplicadas introduzidas pela amplificação e sequenciação PCR causam enviesamentos durante a chamada de pico e análise de enriquecimento. Basepair utiliza a ferramenta Picard para remover as duplicações. Uma vez removidas as duplicatas, deve-se avaliar a Fração Não-Redundante (NRF) das leituras alinhadas. A NRF mede as leituras únicas mapeadas para o genoma de referência. Os experimentos ChIP-seq ideais devem ter menos de três leituras por posição.
Peak Calling
O passo de chamada de pico detecta regiões de interação proteína-DNA enriquecidas no genoma. O gasoduto ChIP-seq da Basepair utiliza o MACS2 para realizar esta análise. No MACS2, a chamada de picos é realizada com base em três etapas principais: estimativa de fragmentos, seguida pela identificação de parâmetros de ruído local e, em seguida, identificação de picos. Como saída desta etapa, os usuários obtêm uma tabela final com informações de pico, como pontuação de enriquecimento, -log10pvalue, -log10qvalue e a posição para início do pico. O uso de amostras de controle é altamente recomendado nesta etapa para comparação com o conjunto de dados alvo investigado. Tenha em mente que bons grupos de controle trazem resultados mais confiáveis.
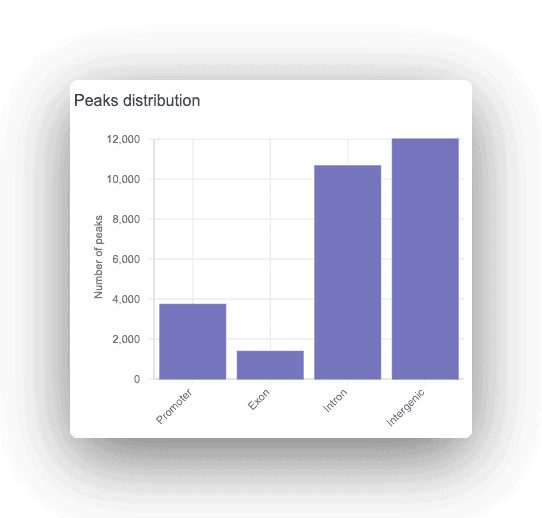
Cada pico é anotado como promotor, intrônico, ou intergênico, com o gene correspondente exibido. Para quaisquer picos encontrados, uma análise do motivo é feita para encontrar locais de ligação de fatores de transcrição super-representados.
Visão geral dos resultados
Um gasoduto ChIP-seq pode fornecer não apenas informações sobre o estado da cromatina, mas também a ligação de fatores de transcrição em um determinado gene ou contexto loci. A ocorrência de modificações de histone e fatores de transcrição em regiões regulatórias de DNA pode constituir uma assinatura epigenética de condição específica. Assim, as perturbações epigenéticas podem ser associadas a fenótipos clínicos. Por exemplo, a heterogeneidade dos estados de cromatina pode levar à resistência ao tratamento do câncer de mama. Estas células tendem a perder marcadores de modificações histônicas repressivas e aumentam ainda mais a expressão de genes conhecidos por promoverem resistência ao tratamento do câncer.
Análise do Bico, Motivo e Caminho na Análise ChIP-Seq Pipeline
A identificação do enriquecimento do fator de transcrição do motivo é utilizada para elucidar se os fatores de transcrição estão cooperando ou competindo em uma determinada região. A identificação de picos nas regiões do motivo do DNA pode melhorar a interpretação dos resultados experimentais. Juntos, tanto as análises de picos como as de motivos dão uma visão do que pode estar ocorrendo dentro de uma célula. A integração de picos e enriquecimento de motivos resulta em uma paisagem epigenômica com possíveis conseqüências biológicas. Além disso, a análise do caminho é usada para identificar proteínas em um caminho. Investigações e conclusões são formuladas com base na presença de uma proteína.
Visualização de dados
Resultados de um pipeline ChIP-seq podem ser visualizados usando um navegador de genoma. Os relatórios Basepair incluem um browser de genoma IGV2 incorporado que lhe permite interagir com os seus dados. Em alternativa, os dados podem ser visualizados através de heatmaps, que são infográficos de intensidade representativa baseados na densidade de dados que mostram a presença ou ausência de marcas específicas. Outros gráficos utilizados aqui incluem o gráfico de enriquecimento, upSet e gráfico de cobertura, que calcula e exibe a cobertura das regiões de pico sobre o genoma.
O browser de genoma é uma excelente ferramenta para visualizar os seus dados genómicos brutos. Está incorporado em todos os relatórios de análise ChIP-seq do Basepair.
1. Grosselin, K., A. Durand, et al. O ChIP-seq de célula única de alto rendimento identifica a heterogeneidade dos estados de cromatina no câncer de mama. Nat Genet, v.51, n.6, Jun, p.1060-1066. 2019.
2. Northrup, D. L. e K. Zhao. Aplicação do ChIP-Seq e técnicas relacionadas ao estudo da função imunológica. Immunity, v.34, n.6, Jun 24, p.830-42. 2011.
3. Park, S. J., J. H. Kim, et al. A ChIP-Seq Data Analysis Pipeline Based on Bioconductor Packages. Genomics Inform, v.15, n.1, Mar, p.11-18. 2017.
4. Pepke, S., B. Wold, et al. Computation for ChIP-seq and RNA-seq studies. Nat Methods, v.6, n.11 Suppl, Nov, p.S22-32. 2009.
5. Satoh, J., N. Kawana, et al. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul Syst Bio, v.7, p.139-52. 2013.