
Introdução
Hypokalemia é um problema clínico comum na prática de endocrinologistas e nefrologistas. Existem muitas causas óbvias de hipocalemia, como diarreia, vômitos ou abuso de diuréticos. Outras causas como as tubulopatias são raramente observadas e o seu diagnóstico é mais desafiador. Existem muitas tubulopatias herdadas e adquiridas causando hipocalemia, por vezes grave e com risco de vida.1
Uma causa relativamente comum mas negligenciada de hipocalemia é a síndrome de Gitelman (GS).2 É uma tubulopatia recessiva com perda de sal causada pela mutação do gene SLC12A3. O gene SLC12A3 codifica o NCCT (co-transportador de cloreto de sódio) do transportador sensível a tiazida. O NCCT está localizado nas células tubulares convolutas distais (CCD), que são responsáveis por 7-10% da absorção tubular eletrolítica.3
As anormalidades laboratoriais mais graves encontradas no GS são hipocalemia e hipomagnesaemia causada pelo desperdício de K+ e Mg2+ renais. Outras alterações típicas são alcalose metabólica, hipocalciúria e hiperaldosteronismo hiperreninêmico.4 Hipofosfatemia leve a moderada é freqüentemente observada.5 Hipofosfatemia grave com hiponatremia grave também foi relatada.6,7
Os primeiros sintomas de GS ocorrem em crianças ou adultos jovens com crescimento normal e histórico de comportamentos de desejo de sal (crianças ansiosas para consumir picles ou salmoura, pepinos salgados, laranjas e limões, crianças lambendo sal de batatas fritas, etc.).8 A apresentação clínica varia entre os pacientes. Alguns são assintomáticos, mas outros desenvolvem complicações que ameaçam a vida. Os homens manifestam um fenótipo mais grave do que as mulheres.8 Os sintomas mais comuns são cãibras e fraqueza muscular, constipação, noctúria, poliúria, sede, polidipsia, arritmias cardíacas, parestesias e aumento do apetite por sal. A hipotensão arterial é comum e, em muitos casos, o sintoma mais proeminente, entretanto, no envelhecimento da população GS pode ocorrer hipertensão arterial.8 A correlação entre anormalidades bioquímicas e sintomas não é forte.9 A GS não interfere no humor e nas relações sociais das crianças.9,10 Caso contrário, os sintomas são mais comuns em adultos e podem ter impacto negativo na sua qualidade de vida. Quarenta e cinco por cento dos pacientes com GS consideram seus sintomas como um problema moderado a grande.11 Exaustão extrema, fraqueza muscular, parestesias, fadiga severa e hipotensão estão associadas com redução leve a severa das atividades diárias.9
Prevalência estimada de GS é de 1:40.0008 e a prevalência de heterozigotos é de pelo menos 1% na população européia. Mais de 180 mutações diferentes na SLC12A3 foram descritas até o momento.12
Relatório de caso
Um homem de 26 anos foi internado no hospital devido à incidência de síncope, fraqueza generalizada e muscular e cãibras musculares. A história do paciente revelou um episódio de síncope com nível de potássio 3,16mmol/l. No acompanhamento ambulatorial, foi observada incidência recorrente de hipocalemia (o menor valor 2,6mmol/l). A pressão arterial estava normal 110/80, a frequência cardíaca era de 72 por minuto; não houve alterações no exame físico. Nem achados neurológicos, peso 74kg, altura 178cm. Na admissão ao hospital a avaliação laboratorial mostrou hipocalemia leve (K+ 3,0mmol/l), hipomagnesaemia (Mg2+ 1,36mg/dl), hipocalciúria (
mg/24h), e alcalose metabólica (HCO3- 29,7mmol/l, BE 5,3mmol/l). A função renal foi boa com eGFR>60ml/min. Os estudos de imagem não foram notáveis, assim como o ECG.
Outras investigações confirmaram que a hipocalemia foi causada pelo desperdício de potássio renal. O desperdício de potássio 24-h, gradiente de K+ transtubular (TTKG), excreção de K+ fracionado (FeK) e relação aleatória K/creatinina (K/Cr) foram típicos para hipercaliuria e são mostrados na Tabela 1.13-15 A magnesiuria diária foi de 64mg/24h, com excreção fracionada de magnésio (FeMg) 15%.
>
Os valores diagnósticos para hipocalemia.13-15
| Diagnóstico de valores para a kaliurese inadequada | Patiente | |
|---|---|---|
| Urinário K+ | >20mmol/24h Se soro K+3.5mmol/l |
57mmol/l (soro K+=3.0mmol/l) |
| FeK=×100% | >6,5% If soro K+3.5mmol/l |
12% (soro K+=3,0mmol/l) |
| TTKG= | TTKG>2 If soro K+3.5mmol/l |
7.3 (soro K+=3.0mmol/l) |
| uK/Cr – razão aleatória de urina K+ para creatinina | uK/Cr>15mmol/g If soro K+3.5mmol/l |
67mmol/g (soro K+=3.0mmol/l) |
Apesar da pressão arterial estar normal, foram realizados testes hormonais para excluir a doença de Conn e a TC abdominal, não tendo sido detectadas quaisquer anomalias nas glândulas supra-renais. Hiperaldosteronismo secundário com níveis de aldosterona 289pg/ml (faixa normal 20-180) e renina 205mIU/ml (faixa normal 2,8-39,9) foram típicos para GS.
O paciente foi diagnosticado como GS com base em um fenótipo clínico. Após o diagnóstico, foi introduzido tratamento com suplementos de potássio e magnésio. Ele recebeu 20mmol de cloreto de potássio e 18mmol de carboxilato de pirrolidona de magnésio, com melhora clínica. A história familiar revelou ligeira hipocalemia assintomática na irmã de 35 anos do paciente. Mas as investigações não revelaram hipocaliuria nem hipomagnesaemia. A SG não foi diagnosticada neste caso.
Análise genética
De acordo com a análise genética, o algoritmo proposto pelo gene Nozu16SLC12A3 deve ser testado em pacientes com alcalose metabólica hipocalêmica, com nascimento a termo, peso normal sem nefrocalcinose, com hipocalúria e hipomagnesaemia.
No caso descrito, foram realizados testes genéticos e duas mutações heterozigóticas: c.35c.35>36insA e c.1095+5G>A foram encontrados na transcrição NM_000339.2 do gene SLC12A3 (Fig. 1). Ambas as mutações ainda não se encontram na versão disponível do banco de dados HGMD(r). A primeira mutação também foi encontrada na mãe da paciente e a segunda no pai. Apenas uma das duas mutações identificadas em nosso paciente c.35_36insA foi encontrada em sua irmã.
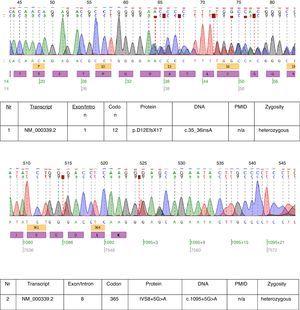
Cromatogramas da sequência de DNA do paciente afectado.
As mutações encontradas neste paciente quase provam o diagnóstico clínico de GS. A relevância patogênica da mutação frameshift é óbvia. Não podemos ter certeza sobre a relevância patogênica da mutação do local de emenda do intron 8. A seqüência de emenda consensual G (100%), T (100%), A (62%), A (68%), G (82%), T (63%) no doador de emenda do intron 8 não corresponde exatamente ao GTgAGc consensual. A mutação encontrada neste paciente desvia ainda mais a sequência do consenso (GtgAac). Além disso, na análise silícica por provador de mutação considera esta mutação como a causa da doença.17
Discussão
Tubulopatias são doenças raras. De acordo com a base de dados RenalTube18 , as tubulopatias primárias mais comuns na população europeia são acidose tubular renal distal, síndrome de Bartter, hipomagnesaemia familiar com hipercalciúria e GS.18 A prevalência de GS é de cerca de 25 casos por 1 milhão.3 Como a GS é uma das mais comuns, provavelmente muitos nefrologistas e endocrinologistas serão confrontados com casos de GS durante suas carreiras. Nosso paciente teve uma apresentação clínica típica de GS e respondeu bem à terapia. O problema com a GS é que a hipocalemia negligenciada pode causar a morte devido a paragem cardíaca ou paralisia dos músculos respiratórios. O sintoma neuromuscular grave, como a paralisia hipocalêmica, ocorre em até 6% dos pacientes (mais comum em pacientes asiáticos).3
As tubulopatias hereditárias têm uma espécie de “imagens-espelho” – tubulopatias adquiridas causadas por diuréticos. Cada diurético (para ser mais preciso: natriuréticos, acquaretics ou glucuréticos) causa anormalidades semelhantes às encontradas nas tubulopatias hereditárias (Tabela 2). Quase em todas as tubulopatias ocorre poliúria e as alterações do nível de potássio estão entre os problemas mais comuns. Em pacientes com GS são encontradas as mutações do gene SLC12A3. Este gene está codificando o transportador sensível a tiazida. Portanto o GS assemelha-se ao tratamento crônico com tiazidas.
Diuréticos e tubulopatias.
| Defeito de um gene único hereditário | Concentração de potássio | Sintomas semelhantes ao uso de: | Gene | Produto do gene | Localização do produto do gene |
|---|---|---|---|---|---|
| Guibaud-Síndrome de Vainsel (RTA 3) | ↓ | Acetazolamida | CA2 | CA II | Tubo proximal |
| Glicosúria renal | – | SGLT2 inhibitorsa (Canagliflozin, Tofogliflozina, Dapagliflozin) | SLC5A2 | Cotransportador de sódio/glucose 2 (SGLT2) | Tubo proximal |
| Síndrome de Bartter | ↓ | Furosemida | SLC12A1b | Na-K-2Cl cotransportador (NKCC2) | Loop of Henle |
| Síndrome de Gitelman | ↓ | Tiazide | SLC12A3 | Tiazide-cotransportador sensível Na-Cl | Tuba distal |
| Diabetes insipidus | – | Vaptan | AVPR2, AQP2 | Receptor V2 | Condutora de recolha |
| Pseudo-hipoaldosteronizme, AD | ← | Spironolactona | NR3C2 | Receptor de mineralocorticóides sensível à espironolactona | Duto colector |
| Pseudo-hipoaldosteronizm, RA | Amiloride | SCNN1A, SCNN1B, SCNN1G | Canal de sódio epitelial sensível à amilorida | Duto colector | |
AR – autossomal recessivo; AD – autossómico dominante.
SGLT2 inibidores (glucuréticos) são os novos medicamentos para o tratamento da diabetes tipo 2; durante o tratamento são observados eventos relacionados à diurese osmótica, como pollakiuria e poliúria.
Embora existam diferentes genes de Bartter, este é o que imita a furosemida.
>Aproximidade à hipocalemia
GS também é conhecida como hipomagnesaemia hipocalémica familiar, porque a hipocalemia é o fenómeno mais comum. Em um paciente com hipocalemia e suspeita de tubulopatia, deve-se confirmar a hipocaliuria. O desperdício renal de potássio pode ser comprovado calculando o TKKG, FeK, relação aleatória K/Cr ou após a coleta de urina 24 horas (Tabela 1). A avaliação da caliuria deve ser feita quando o paciente não está tomando diuréticos ou suplementação de potássio e quando o nível de potássio está baixo. Quando a excreção de potássio está abaixo de 30mmol/l e o TTKG está baixo, a hipocalemia é causada por perda extrarrenal ou um deslocamento de K+ transcelular.
Quando a perda renal excessiva de potássio em paciente com hipocalemia é confirmada, o diagnóstico adicional é baseado no pH e na pressão arterial. Em muitos casos, estes estudos simples serão suficientes para o diagnóstico. Um problema importante de interesse crescente é o aldosteronismo primário (AP). Alguns estudos clínicos têm sugerido que a AF é a causa de mais de 10% da hipertensão arterial (HA), sendo mais comum em pacientes com HA resistentes a agentes anti-hipertensivos.19 A hipocalemia é um dos sintomas “clássicos” da AF, mas não é tão comum como foi considerado.20 Ainda assim, como não há um padrão ouro no diagnóstico da PA, vale a pena lembrar sobre o eixo RAA em pacientes com HA e hipocalemia.19
Diagnóstico de hipomagnesemia
Hipomagnesemia é uma anormalidade comum em pacientes com GS, embora não seja observada em todos os casos.21 Em pacientes normomagnesêmicos com GS, as manifestações clínicas e anormalidades eletrolíticas são mais leves.22
Semelhante aos íons potássio, os de magnésio são filtrados livremente pelos glomérulos. 10% do Mg2+ filtrado é absorvido pelo túbulo proximal, 50-70% no membro ascendente do laço de Henle. A reabsorção distal depende dos canais epiteliais Mg2+ TRPM6. Magnesiuria depende da ingestão oral e FeMg deve ser calculado para estabelecer o desperdício renal de Mg2+ (FeMg=(urina magnésio× creatinina sérica)/×100). FeMg inferior a 2% sugere má ingestão, perdas GI ou deslocamento de Mg em células. FeMg acima de 4% sugere desperdício de Mg2+ renal. Quando a perda de Mg renal é comprovada, deve-se calcular a razão aleatória Ca/Cr. A razão Urina Ca/Cr 0,3 é típica na hipermagesúria na síndrome de Bartter, assim como hipomagnesaemia recessiva isolada com normocalciúria, hipomagnesaemia familiar com hipercalciúria e nefrocalcinose, hipocalcemia autossômica dominante com hipercalciúria, tratamento com diuréticos de laço e nefropatia causada por algumas nefrotoxinas.23
Magnésio e homeostase de potássio estão relacionados entre si, e a depleção de potássio não pode ser corrigida até a correção da hipomagnesaemia.23 Em média, os pacientes devem receber até 500mEq de potássio, 4-5mg/kg/dia de 5-10mg de cloreto de magnésio. Amilorida (5-10mg/dia) e espironolacton (200-300mg) são úteis.3
Os sintomas clínicos e os resultados laboratoriais são essenciais para o diagnóstico. Em casos com fenótipo típico, alguns autores recomendam a realização de um teste de tiazida para confirmar o diagnóstico.24 Em casos apresentados, a investigação genética revelou duas novas mutações do gene SLC12A3. Elas são provavelmente causadas pela falta de mutações funcionais do TCCN.
Conclusões
SG é uma das raras causas de hipocalemia, e parece ser um desafio para os médicos. Mostramos neste trabalho que se alguém se lembrar de uma abordagem muito simples da hipocalemia e estiver ciente da ação dos diuréticos e sua semelhança com as tubulopatias hereditárias, o diagnóstico pode ser bastante simples. A SG deve ser diferenciada de outras tubulopatias (herdadas e adquiridas) e de outras causas de hipocalemia (por exemplo, a doença de Conn). A história familiar pode revelar pacientes assintomáticos com GS. O tratamento adequado protege os pacientes de complicações potencialmente perigosas.