
Breve introduzione al ChIP-Seq

Le interazioni proteine-DNA sono ampiamente utilizzate per chiarire i meccanismi alla base della fisiologia cellulare. Lo sviluppo della tecnologia di immunoprecipitazione della cromatina (ChIP) ha permesso lo studio di tali meccanismi. Dopo un ulteriore sviluppo, è nata un’alternativa di sequenziamento profondo (ChiP-Seq), che offre vantaggi in termini di specificità e sensibilità.
Un esperimento ChIP-Seq inizia con un cross-link di cellule intere con formaldeide, seguito da sonicazione e isolamento del DNA. Dopo di che, viene eseguita l’immunoprecipitazione del complesso DNA-proteina, che consiste in anticorpi che si legano a proteine specifiche. Gli immunocomplessi formati vengono precipitati e purificati. Infine, il DNA viene sequenziato, generando dati ad alta risoluzione dei siti arricchiti. Questo approccio, insieme a una pipeline ChIP-seq ben consolidata, permette ai ricercatori di catturare i fattori di trascrizione del DNA, i siti di modifica degli istoni, le alterazioni epigenetiche e le firme della rete di regolazione genica.
Rilevanza clinica e applicazioni
Gli squilibri epigenetici nelle malattie e nelle condizioni di salute possono coinvolgere la modifica degli istoni e i fattori di trascrizione alterati. Qui, gli studi ChIP-Seq sono stati utilizzati per chiarire i meccanismi molecolari patologici alla base del cancro e di altre malattie. L’analisi ChIP-seq contribuisce anche alla comprensione del ruolo dei fattori di trascrizione durante le malattie. Infatti, alcune trascrizioni sembrano essere alterate durante le manifestazioni del fenotipo clinico.
Panoramica della pipeline ChIP-Seq
La pipeline di analisi ChIP-Seq è il componente principale dei progetti di interazione DNA-proteina e consiste di diversi passaggi, tra cui l’elaborazione dei dati grezzi, l’analisi del controllo di qualità, l’allineamento al genoma di riferimento, il controllo di qualità delle letture allineate, la chiamata dei picchi, l’annotazione e la visualizzazione. Tuttavia, avere un disegno sperimentale ben studiato è fondamentale per ottenere risultati di alta qualità in un esperimento ChIP-seq. Prima di iniziare l’analisi, è essenziale considerare parametri come le repliche dei campioni, i gruppi di controllo, i kit di sequenziamento e le piattaforme di sequenziamento.
Controllo di qualità
Tutti i rapporti Basepair forniscono punteggi di qualità per aiutare a scoprire potenziali problemi di sequenziamento o di contaminazione nei tuoi dati di input.
La fase di controllo di qualità (QC) mira a valutare la qualità dei dati ad alta produttività generati dal sequenziamento. Questo passo è simile a quelli eseguiti nelle analisi DNA-seq e RNA-seq. Qui, le principali metriche valutate includono la qualità della sequenza e della base, il contenuto di GC, la presenza di adattatori di sequenziamento e le sequenze sovrarappresentate. Uno dei programmi più comunemente usati per questo tipo di analisi è FastQC. Inoltre, se vengono identificate sequenze di bassa qualità, queste possono essere successivamente rimosse durante la fase di trimming. Sebbene sia un passo opzionale, il trimming migliora la qualità dei dati conservando solo letture di alta qualità.
Allineamento
Dopo la misurazione QC, le letture ChIP-Seq vengono allineate a un genoma di riferimento. La mappatura delle letture permette ai ricercatori di identificare l’origine di una sequenza di lettura nel genoma. Gli strumenti software di allineamento più utilizzati includono Bowtie e BWA, entrambi utilizzati nelle pipeline ChIP-seq di Basepair. Entrambi gli strumenti mappano le sequenze a bassa divergenza rispetto a un genoma di riferimento.
Il flusso del conteggio delle letture aiuta a fornire una panoramica generale delle letture utilizzabili alla fine dei processi di taglio, allineamento e deduplicazione. Pensa alla figura come una catena di montaggio per l’analisi dei dati: dati grezzi in ingresso, ottieni un output di letture utilizzabili.
Controllo di qualità delle letture allineate
Il passo successivo consiste nell’inferenza QC del dataset allineato. Durante il processo di mappatura, i duplicati di lettura introdotti dall’amplificazione della PCR e dal sequenziamento causano distorsioni durante la chiamata dei picchi e l’analisi di arricchimento. Basepair utilizza lo strumento Picard per rimuovere i duplicati. Una volta rimossi i duplicati, dovresti valutare la Non-Redundant Fraction (NRF) delle letture allineate. NRF misura le letture uniche che mappano il genoma di riferimento. Gli esperimenti ChIP-seq ideali dovrebbero avere meno di tre letture per posizione.
Peak Calling
La fase di peak calling rileva le regioni di interazione proteina-DNA arricchite sul genoma. La pipeline ChIP-seq di Basepair utilizza MACS2 per eseguire questa analisi. In MACS2, la chiamata dei picchi viene eseguita sulla base di tre fasi principali: stima dei frammenti, seguita dall’identificazione dei parametri di rumore locale e quindi dall’identificazione dei picchi. Come output di questa fase, gli utenti ottengono una tabella finale con informazioni sui picchi, come il punteggio di arricchimento, il -log10pvalue, il -log10qvalue e la posizione all’inizio del picco. L’uso di campioni di controllo è altamente raccomandato in questo passaggio per il confronto con il set di dati di destinazione studiato. Tenete a mente che buoni gruppi di controllo portano a risultati più affidabili.
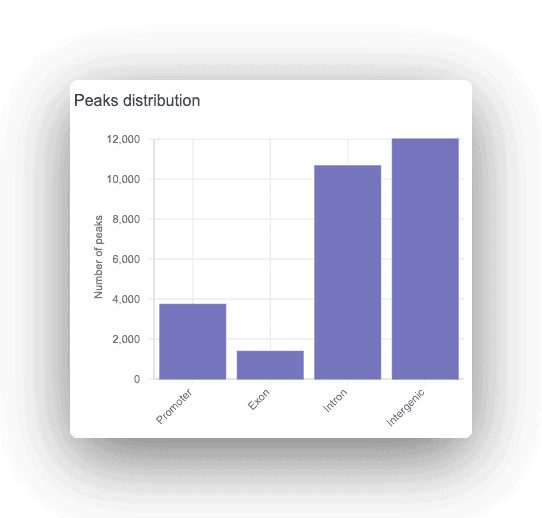
Ogni picco viene annotato come promotore, intronico o intergenico, con il gene corrispondente visualizzato. Per ogni picco trovato, viene fatta un’analisi dei motivi per trovare i siti di legame dei fattori di trascrizione sovrarappresentati.
Panoramica dei risultati
Una pipeline ChIP-seq può fornire non solo informazioni sullo stato della cromatina ma anche sul legame dei fattori di trascrizione in un determinato gene o contesto di loci. La presenza di modifiche degli istoni e dei fattori di trascrizione nelle regioni di regolazione del DNA può costituire una firma epigenetica condizione-specifica. Così, le perturbazioni epigenetiche possono essere associate a fenotipi clinici. Per esempio, l’eterogeneità degli stati della cromatina può portare alla resistenza al trattamento nel cancro al seno. Queste cellule tendono a perdere i marcatori repressivi delle modifiche degli istoni e ad aumentare ulteriormente l’espressione dei geni noti per promuovere la resistenza al trattamento del cancro.
Peak, Motif and Pathway Analysis in ChIP-Seq Analysis Pipeline
L’identificazione dell’arricchimento dei fattori di trascrizione motif è usata per chiarire se i fattori di trascrizione stanno cooperando o competendo in una data regione. L’identificazione dei picchi nelle regioni del motivo del DNA può migliorare l’interpretazione dei risultati sperimentali. Insieme, sia i picchi che le analisi dei motivi danno un’idea di ciò che può accadere all’interno di una cellula. L’integrazione degli arricchimenti dei picchi e dei motivi danno come risultato un paesaggio epigenomico con possibili conseguenze biologiche. Inoltre, l’analisi dei percorsi è usata per identificare le proteine in un percorso. Le indagini e le conclusioni sono formulate sulla base della presenza di una proteina.
Visualizzazione dei dati
I dati risultanti da una pipeline ChIP-seq possono essere visualizzati utilizzando un genome browser. I report Basepair includono un browser del genoma IGV2 incorporato che ti permette di interagire con i tuoi dati. I dati possono in alternativa essere visualizzati utilizzando heatmap, che sono infografiche di intensità rappresentativa basate sulla densità dei dati che mostrano la presenza o l’assenza di segni specifici. Altri grafici usati qui includono plot di arricchimento, upSet e plot di copertura, che calcola e visualizza la copertura delle regioni di picco sul genoma.
Il browser del genoma è un ottimo strumento per visualizzare i vostri dati genomici grezzi. È integrato in ogni rapporto di analisi ChIP-seq su Basepair.
1. Grosselin, K., A. Durand, et al. High-throughput single-cell ChIP-seq identifica l’eterogeneità degli stati della cromatina nel cancro al seno. Nat Genet, v.51, n.6, giugno, p.1060-1066. 2019.
2. Northrup, D. L. e K. Zhao. Applicazione di ChIP-Seq e tecniche correlate allo studio della funzione immunitaria. Immunità, v.34, n.6, Jun 24, p.830-42. 2011.
3. Park, S. J., J. H. Kim, et al. Un ChIP-Seq Data Analysis Pipeline basato su pacchetti Bioconductor. Genomics Inform, v.15, n.1, Mar, p.11-18. 2017.
4. Pepke, S., B. Wold, et al. Calcolo per ChIP-seq e RNA-seq studi. Nat Methods, v.6, n.11 Suppl, Nov, p.S22-32. 2009.
5. Satoh, J., N. Kawana, et al. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul Syst Bio, v.7, p.139-52. 2013.