
Nota dell’editore: Gli articoli Platinum Highlight sono pubblicazioni degne di nota selezionate periodicamente dal Dr. Craig Reynolds, direttore associato, National Cancer Institute, tra le pubblicazioni Platinum di più recente pubblicazione.![]()
Quando Alison Rattray e i colleghi del Gene Regulation and Chromosome Biology Laboratory (GRCBL) hanno esaminato una cellula di lievito mutante che avevano isolato in uno schermo, hanno notato qualcosa di strano.
Il DNA mostrava un “riordinamento molto specifico, ma strano”, ha spiegato. La disposizione si è rivelata essere un palindromo del DNA, “aprendo la porta allo studio di questi motivi elusivi del DNA”, ha detto.
Scienziato del GRCBL, NCI Center for Cancer Research, Rattray ha detto che, mentre la loro scoperta è avvenuta un certo numero di anni fa, il loro gruppo continua a studiare la riparazione del DNA e i riarrangiamenti che risultano in una riparazione anormale “a causa dell’associazione con alcuni tumori”. L’obiettivo del gruppo è quello di capire meglio l’origine di questi riarrangiamenti, ha aggiunto.
Rattray è uno dei principali contributori alla ricerca riportata in BMC Genomics che descrive il metodo recentemente sviluppato dal gruppo per il sequenziamento genomico dei palindromi del DNA in una linea cellulare di cancro.
Cos’è un palindromo del DNA?
Una sequenza palindromica di nucleotidi (che sono etichettati A, T, C, o G) si verifica quando i filamenti complementari di DNA leggono lo stesso in entrambe le direzioni, sia dall’estremità 5-prima che dall’estremità 3-prima. Per esempio, la sequenza GGATCC su un filamento di DNA è considerata un palindromo perché la sequenza sul suo filamento complementare è CCTAGG.
A causa della complementarità della sequenza, le sequenze palindromiche possono anche ripiegarsi su se stesse, formando anelli a forcina o cruciformi che sono estrusi dalla normale doppia elica, ha detto Rattray. “Piccole forcine non sono problematiche, ma quando i palindromi sono lunghi (più di 100 paia di basi), interferiscono con i normali processi cellulari come la trascrizione e la replicazione”, ha spiegato.
Alcune cellule tumorali mostrano massicci riarrangiamenti del genoma, che includono amplificazioni geniche, traslocazioni e delezioni, e questi riarrangiamenti sono spesso associati alla presenza di un palindromo, suggerendo una possibile correlazione tra il palindromo e i riarrangiamenti genici. Come questi eventi si verifichino non è ben compreso. Ciò che è noto, tuttavia, è che tali riarrangiamenti sono associati alla progressione e alla prognosi del cancro, ha detto Rattray.
Nuova ipotesi per il riarrangiamento genomico
Secondo Rattray, il modello preferito, originariamente proposto più di 60 anni fa da Barbara McClintock, Ph.D., suggerisce che, dopo una rottura cromosomica, i cromatidi fratelli si replicano e si fondono, creando un cromosoma con due centromeri uniti da un palindromo del DNA. Nel modello McClintock, avere due centromeri porta a ulteriori riarrangiamenti genomici.
Tuttavia, Rattray ha detto che il suo gruppo e altri hanno dimostrato che “i palindromi del DNA sono instabili e possono portare a riarrangiamenti del genoma da soli, suggerendo ulteriormente che i palindromi potrebbero sorgere non solo dalla fusione dei cromatidi sorella, ma anche da altri meccanismi, come gli errori di replicazione.”
Il gruppo ha ipotizzato che “nei tumori che subiscono riarrangiamenti massicci, le cellule sono suscettibili alla formazione di palindromi, e una volta formati, l’instabilità del palindromo porta a ulteriori riarrangiamenti, tra cui l’amplificazione genica, traslocazioni e delezioni”, ha detto Rattray. “Qualsiasi riarrangiamento genico è mutageno, e i riarrangiamenti che promuovono la crescita cellulare, come nel cancro, saranno naturalmente favoriti dalla selezione”.
Nuova tecnologia identifica e caratterizza i palindromi
I ricercatori hanno sviluppato una tecnologia che permetterà loro di esaminare i tumori, con l’obiettivo di capire la probabilità di formazione di palindromi in questi tumori, ha detto Rattray. Sperano di imparare quali eventi iniziano tali formazioni instabili, e questa nuova comprensione potrebbe portare a nuovi trattamenti. Per esempio, ha detto, il gruppo ha già determinato che alcune cellule di lievito che sono suscettibili alla formazione di palindromi sono molto più sensibili delle cellule normali alle radiazioni così come ai composti spesso utilizzati nel trattamento del cancro, come il cisplatino.
“Attualmente, sto cercando di stabilire metodi per arricchire selettivamente palindromi dal resto del DNA cellulare, che permetterà una maggiore sensibilità nell’analisi del contenuto palindromo delle cellule tumorali,” ha detto. Nel metodo precedente, i ricercatori hanno perso le sequenze di giunzione che potrebbero fornire indizi sull’origine dei palindromi, e hanno dovuto analizzarli uno per uno, ha spiegato. “Abbiamo ora dimostrato che la piattaforma PacBio può facilmente sequenziare attraverso un palindromo del DNA” ha detto.
Rattray ha ricevuto il suo dottorato presso l’Università di Washington, a Seattle, dove ha studiato la replicazione retrovirale. Dopo una borsa di studio post-dottorato alla Columbia University, dove ha studiato la ricombinazione del DNA e i riarrangiamenti indotti dalle rotture del doppio filamento del DNA nel lievito, si è unita al NCI a Frederick, lavorando nel laboratorio di Jeffrey Strathern, Ph.D., capo del GRCBL.
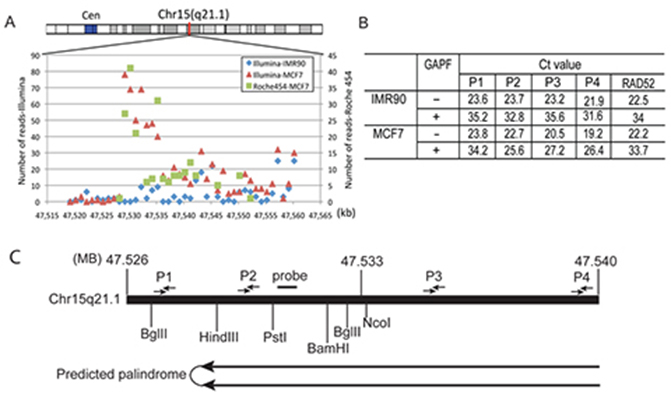
Strategia di mappatura del palindromo. (A) Distribuzione della densità di lettura nel Chr15q21.1: 47,529,204-47,550,373 regione mostrata come 1-kb bins. (B) analisi qPCR per monitorare l’arricchimento del palindromo e determinare la direzionalità del palindromo Chr15q21.1. I ricercatori hanno calcolato la quantità di deplezione di una regione specifica TaqMan primer set basato sul valore Ct prima e dopo il protocollo GAPF in entrambi i campioni IMR-90 e MCF-7. L’arricchimento delle pieghe si basa sul confronto dell’impoverimento delle pieghe tra i diversi set di primer (P1, P2, P3 e P4) rispetto a una sequenza a copia unica nel genoma (RAD52). Le posizioni dei set di primer TaqMan P1, P2, P3 e P4 sono indicate in (C), mappa della regione genomica Chr15: 47.520.000-47.550.000 con siti di restrizione e posizioni dei primer. Figura da Yang et al., GAP-Seq: un metodo per l’identificazione di palindromi del DNA, BMC Genomics 2014, 15:394; doi:10.1186/1471-2164-15-394.