
Introduzione
L’ipokaliemia è un problema clinico comune nella pratica degli endocrinologi e dei nefrologi. Ci sono molte cause ovvie di ipopotassiemia come diarrea, vomito o abuso di diuretici. Altre cause come le tubulopatie sono raramente osservate e la loro diagnosi è più impegnativa. Ci sono molte tubulopatie ereditate e acquisite che causano ipopotassiemia, a volte grave e pericolosa per la vita.1
Una causa relativamente comune ma trascurata di ipopotassiemia è la sindrome di Gitelman (GS).2 È una tubulopatia recessiva che perde sale causata dalla mutazione del gene SLC12A3. Il gene SLC12A3 codifica il trasportatore sensibile ai tiazidici NCCT (co-trasportatore di cloruro di sodio). NCCT si trova nelle cellule tubulari convolute distali (DCC), che sono responsabili del 7-10% dell’assorbimento tubulare degli elettroliti.3
Le anomalie di laboratorio più gravi riscontrate nella GS sono l’ipopotassiemia e l’ipomagnesiemia causate dalla perdita renale di K+ e Mg2+. Altri cambiamenti tipici sono l’alcalosi metabolica, l’ipocalciuria e l’iperaldosteronismo iperreninemico.4 L’ipofosfatemia da lieve a moderata è frequentemente osservata.5 È stata riportata anche una grave ipofosfatemia con grave iponatriemia.6,7
I primi sintomi della GS si verificano nei bambini o nei giovani adulti con crescita normale e con una storia di comportamenti di brama di sale (bambini desiderosi di consumare sottaceti o salamoia, cetrioli salati, arance e limoni, bambini che leccano il sale dalle patatine, ecc. Alcuni sono asintomatici, ma altri sviluppano complicazioni pericolose per la vita. I maschi manifestano un fenotipo più grave rispetto alle femmine.8 I sintomi più comuni sono crampi muscolari e debolezza, stipsi, nicturia, poliuria, sete, polidipsia, aritmie cardiache, parestesie e aumento dell’appetito di sale. L’ipotensione arteriosa è comune e in molti casi il sintomo più importante, tuttavia, nella popolazione anziana con GS può verificarsi ipertensione.8 La correlazione tra anomalie biochimiche e sintomi non è forte.9 La GS non interferisce con l’umore e le relazioni sociali dei bambini.9,10 Altrimenti i sintomi sono più comuni negli adulti e possono avere un impatto negativo sulla loro qualità di vita. Il quarantacinque per cento dei pazienti con GS considera i propri sintomi come un problema da moderato a grande.11 Estrema spossatezza, debolezza muscolare, parestesie, grave affaticamento e ipotensione sono associati a una riduzione da lieve a grave delle attività quotidiane.9
La prevalenza stimata della GS è di 1:40.0008 e la prevalenza degli eterozigoti è almeno dell’1% nella popolazione europea. Più di 180 diverse mutazioni in SLC12A3 sono state descritte fino ad ora.12
Rapporto del caso
Un uomo di 26 anni è stato ricoverato in ospedale per incidenza di sincope, debolezza generalizzata e muscolare e crampi muscolari. L’anamnesi del paziente ha rivelato un episodio di sincope con un livello di potassio di 3,16 mmol/l. In un ulteriore follow-up nella valutazione ambulatoriale, è stata osservata un’incidenza ricorrente di ipokaliemia (il valore più basso 2,6mmol/l). La pressione sanguigna era normale 110/80, la frequenza cardiaca era di 72 al minuto; non c’erano cambiamenti nell’esame fisico. Né risultati neurologici, peso 74kg, altezza 178cm. All’ammissione in ospedale la valutazione di laboratorio ha mostrato una lieve ipokaliemia (K+ 3.0mmol/l), ipomagnesiemia (Mg2+ 1.36mg/dl), ipocalciuria (
mg/24h), e alcalosi metabolica (HCO3- 29.7mmol/l, BE 5.3mmol/l). La funzione renale era buona con eGFR>60ml/min. Gli studi di imaging erano irrilevanti, così come l’ECG.
Indagini ulteriori hanno confermato che l’ipokaliemia era causata dal deperimento renale del potassio. Lo spreco di potassio nelle 24 ore, il gradiente K+ transtubulare (TTKG), l’escrezione frazionata di K+ (FeK) e il rapporto K/creatinina casuale (K/Cr) erano tipici dell’iperkaliuria e sono riportati nella tabella 1.13-15 La magnesiuria giornaliera era di 64mg/24h, con escrezione frazionale di magnesio (FeMg) del 15%.
I valori diagnostici per ipokaliemia.13-15
| Valori diagnostici per una kaliuresi inappropriata | Paziente | |
|---|---|---|
| K+ urinario | >20mmol/24h Se K+3 siero.5mmol/l |
57mmol/l (siero K+=3.0mmol/l) |
| FeK=×100% | >6,5% Se K+3.5mmol/l |
12% (siero K+=3.0mmol/l) |
| TTKG= | TTKG>2 Se K+3.5mmol/l |
7.3 (siero K+=3.0mmol/l) |
| uK/Cr – rapporto casuale tra K+ e creatinina nelle urine | uK/Cr>15mmol/g Se K+3.5mmol/l |
67mmol/g (siero K+=3.0mmol/l) |
Anche se la pressione sanguigna era normale, i test ormonali per escludere la malattia di Conn e la TAC addominale sono stati eseguiti e non hanno rivelato alcuna anomalia nelle ghiandole surrenali. L’iperaldosteronismo secondario con livelli di aldosterone 289pg/ml (range normale 20-180) e di renina 205mIU/ml (range normale 2,8-39,9) era tipico della GS.
Il paziente è stato diagnosticato come GS sulla base del fenotipo clinico. Dopo la diagnosi, è stato introdotto un trattamento con integratori di potassio e magnesio. Ha ricevuto 20mmol di cloruro di potassio e 18mmol di pirrolidone carbossilato di magnesio, con miglioramento clinico. L’anamnesi familiare ha rivelato una lieve ipokaliemia asintomatica nella sorella 35enne del paziente. Ma le indagini non hanno mostrato né iperkaliuria né ipomagnesiemia. La GS non è stata diagnosticata in questo caso.
Analisi genetica
Secondo l’analisi genetica, algoritmo proposto da Nozu16Il gene SLC12A3 dovrebbe essere testato in pazienti con alcalosi metabolica ipokaliemica, con nascita a termine, peso normale senza nefrocalcinosi, con ipocalcuria e ipomagnesiemia.
Nel caso descritto è stato eseguito il test genetico e due mutazioni eterozigoti: c.35_36insA e c.1095+5G>A sono state trovate nel trascritto NM_000339.2 del gene SLC12A3 (Fig. 1). Entrambe le mutazioni non sono ancora presenti nella versione disponibile del database HGMD(r). La prima mutazione è stata trovata anche nella madre del paziente e la seconda nel padre. Solo una delle due mutazioni identificate nel nostro paziente c.35_36insA è stata trovata in sua sorella.
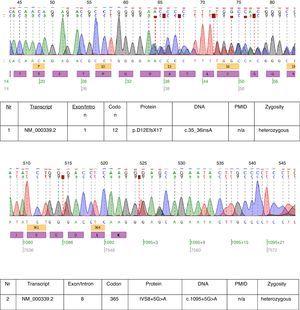
Cromatogrammi del sequenziamento del DNA del paziente affetto.
Le mutazioni trovate in questo paziente provano quasi la diagnosi clinica di GS. La rilevanza patogenetica della mutazione frameshift è ovvia. Non possiamo essere sicuri della rilevanza patogenetica della mutazione del sito di splice dell’introne 8. La sequenza di consenso di splice G (100%), T (100%), A (62%), A (68%), G (82%), T (63%) al donatore di splice dell’introne 8 non corrisponde esattamente al consenso GTgAGc. La mutazione trovata in questo paziente devia ulteriormente la sequenza dal consenso (GtgAac). Inoltre, l’analisi in silico di mutation taster considera questa mutazione come causa della malattia.17
Discussione
Le tubulopatie sono malattie rare. Secondo il database RenalTube18 le tubulopatie primarie più comuni nella popolazione europea sono l’acidosi tubulare renale distale, la sindrome di Bartter, l’ipomagnesiemia familiare con ipercalciuria e la GS.18 La prevalenza della GS è di circa 25 casi su 1 milione.3 Poiché la GS è una delle più comuni, probabilmente molti nefrologi ed endocrinologi si troveranno di fronte a casi di GS durante la loro carriera. Il nostro paziente aveva una presentazione clinica tipica della GS e rispondeva bene alla terapia. Il problema della GS è che l’ipokaliemia trascurata potrebbe causare la morte per arresto cardiaco o paralisi dei muscoli respiratori. Il sintomo neuromuscolare grave come la paralisi ipokaliemica si verifica fino al 6% dei pazienti (più comune nei pazienti asiatici).3
Le tubulopatie ereditate hanno una sorta di “immagini speculari” – tubulopatie acquisite causate da diuretici. Tutti i diuretici (per essere precisi: natriuretici, acquaretici o glucuretici) causano anomalie simili a quelle riscontrate nelle tubulopatie ereditarie (tabella 2). Quasi in ogni tubulopatia si verifica poliuria, e le alterazioni del livello di potassio sono tra i problemi più comuni. Nei pazienti con GS si trovano le mutazioni del gene SLC12A3. Questo gene codifica il trasportatore sensibile ai tiazidi. Pertanto la GS assomiglia al trattamento cronico con tiazidi.
Diuretici e tubulopatie.
| Deficienza ereditaria di un singolo gene | Concentrazione di potassio | Sintomi simili all’uso di: | Gene | Prodotto genico | Localizzazione del prodotto genico |
|---|---|---|---|---|---|
| Guibaud-Vainsel sindrome (RTA 3) | ↓ | Acetazolamide | CA2 | CA II | Tubulo prossimale |
| Glicosuria renale | – | Inibitori SGLT2orsa (Canagliflozin, Tofogliflozin, Dapagliflozin) | SLC5A2 | Cotrasportatore sodio/glucosio 2 (SGLT2) | Tubulo prossimale |
| Sindrome di Bartter | ↓ | Furosemide | SLC12A1b | Na-K-2Cl cotrasportatore (NKCC2) | Ciclo di Henle |
| Sindrome di Gitelman | ↓ | Tiazide | SLC12A3 | Tiazide-sensibile Na-Cl cotrasportatore | Tubulo distale |
| Diabete insipido | – | Vaptan | AVPR2, AQP2 | Ricettore V2 | Dotto collettore |
| Pseudoipoaldosteronizm, AD | ← | Spironolattone | NR3C2 | Ricettore dei mineralocorticoidi sensibile allo spironolattone | Dotto collettore |
| Pseudohypoaldosteronizm, AR | Amiloride | SCNN1A, SCNN1B, SCNN1G | Canale del sodio epiteliale sensibile all’amiloride | Dotto collettore | |
AR – autosomico recessivo; AD – autosomico dominante.
Gli inibitori di SGLT2 (glucuretici) sono i nuovi farmaci per la gestione del diabete di tipo 2; durante il trattamento si osservano eventi legati alla diuresi osmotica come pollakiuria e poliuria.
Anche se ci sono diversi geni Bartter questo è quello che imita il furosemide.
Approccio all’ipokaliemia
La GS è anche conosciuta come ipomagnesiemia ipokaliemica familiare, perché l’ipokaliemia è il fenomeno più comune. In un paziente con ipokaliemia e sospetto di tubulopatia si deve confermare l’iperkaliuria. Il deperimento renale del potassio può essere dimostrato calcolando il TKKG, il FeK, il rapporto K/Cr casuale o dopo una raccolta di urine di 24 ore (tabella 1). La valutazione della kaliuria dovrebbe essere eseguita quando il paziente non assume né diuretici né supplementi di potassio e quando il livello di potassio è basso. Quando l’escrezione di potassio è inferiore a 30mmol/l e il TTKG è basso, l’ipokaliemia è causata da una perdita extrarenale o da uno spostamento transcellulare di K+.
Quando viene confermata un’eccessiva perdita renale di potassio nel paziente con ipokaliemia, un’ulteriore diagnosi si basa su pH e pressione sanguigna. In molti casi questi semplici studi saranno sufficienti per la diagnosi. Un importante problema di crescente interesse è l’aldosteronismo primario (PA). Alcuni studi clinici hanno suggerito che il PA è la causa di oltre il 10% dell’ipertensione arteriosa (AH), ed è più comune nei pazienti con AH resistenti agli agenti antipertensivi.19 L’ipopotassiemia è uno dei sintomi “classici” del PA, ma non è così comune come è stato considerato.20 Tuttavia, poiché non esiste un gold standard nella diagnosi di AP, vale la pena ricordare l’asse RAA nei pazienti con AH e ipopotassiemia.19
Diagnosi di ipomagnesiemia
L’ipomagnesiemia è un’anomalia comune nei pazienti con GS, anche se non si osserva in tutti i casi.21 Nei pazienti normomagnesemici con GS, le manifestazioni cliniche e le anomalie elettrolitiche sono più lievi.22
Similmente agli ioni potassio, quelli di magnesio sono filtrati liberamente dai glomeruli. Il 10% del Mg2+ filtrato è assorbito dal tubulo prossimale, il 50-70% nell’arto ascendente dell’ansa di Henle. Il riassorbimento distale dipende dai canali epiteliali Mg2+ TRPM6. La magnesiuria dipende dall’assunzione orale e il FeMg deve essere calcolato per stabilire il deficit di Mg2+ renale (FeMg=(magnesio urinario×creatinina sierica)/×100). FeMg inferiore al 2% suggerisce una scarsa assunzione, perdite GI o spostamento di Mg nelle cellule. FeMg superiore al 4% suggerisce uno spreco renale di Mg2+. Quando lo spreco renale di Mg è provato, si dovrebbe calcolare il rapporto Ca/Cr casuale. Il rapporto Ca/Cr delle urine 0,3 è tipico dell’ipermagesuria nella sindrome di Bartter, così come l’ipomagnesiemia recessiva isolata con normocalciuria, l’ipomagnesiemia familiare con ipercalciuria e nefrocalcinosi, l’ipocalcemia autosomica dominante con ipercalciuria, il trattamento con diuretici dell’ansa e la nefropatia causata da alcune nefrotossine.23
L’omeostasi del magnesio e del potassio sono legate l’una all’altra, e la deplezione di potassio non può essere corretta fino alla correzione dell’ipomagnesiemia.23 In media i pazienti dovrebbero ricevere fino a 500mEq di potassio, 4-5mg/kg/giorno di 5-10mg di cloruro di magnesio. L’amiloride (5-10mg/giorno) e lo spironolattone (200-300mg) sono utili.3
I sintomi clinici e i risultati di laboratorio sono essenziali per la diagnosi. Nei casi con fenotipo tipico alcuni autori raccomandano di eseguire un test tiazidico per confermare la diagnosi.24 Nel caso presentato l’indagine genetica ha rivelato due nuove mutazioni del gene SLC12A3. Essi sono probabilmente causati dalla mancanza di mutazioni funzionali di NCCT.
Conclusioni
La sindromeGS è una delle cause rare di ipopotassiemia, e sembra una sfida per i medici. Mostriamo in questo articolo che se ci si ricorda di un approccio molto semplice all’ipopotassiemia e si conosce l’azione dei diuretici e la loro somiglianza con le tubulopatie ereditarie, la diagnosi potrebbe essere abbastanza semplice. La GS deve essere differenziata da altre tubulopatie (ereditarie e acquisite) e da altre cause di ipopotassiemia (per esempio la malattia di Conn). L’anamnesi familiare può rivelare pazienti asintomatici con GS. Un trattamento adeguato protegge i pazienti da complicazioni potenzialmente pericolose.