
Introductie
Hypokaliëmie is een veelvoorkomend klinisch probleem in de praktijk van endocrinologen en nefrologen. Er zijn veel voor de hand liggende oorzaken van hypokaliëmie zoals diarree, braken of misbruik van diuretica. Andere oorzaken zoals tubulopathieën worden zelden waargenomen en hun diagnose is een grotere uitdaging. Er zijn vele erfelijke en verworven tubulopathieën die hypokaliëmie veroorzaken, soms ernstig en levensbedreigend.1
Een relatief vaak voorkomende maar over het hoofd geziene oorzaak van hypokaliëmie is het Gitelman syndroom (GS).2 Het is een recessieve zout-verliezende tubulopathie veroorzaakt door de SLC12A3 genmutatie. Het SLC12A3-gen codeert voor de thiazide-gevoelige transporteur NCCT (natriumchloride co-transporter). NCCT bevindt zich in de distale convoluted tubular cells (DCC), die verantwoordelijk zijn voor 7-10% van de elektrolyt tubulaire absorptie.3
De meest ernstige laboratoriumafwijkingen die bij GS worden gevonden zijn hypokaliëmie en hypomagnesiëmie veroorzaakt door renale K+ en Mg2+ verspilling. Andere typische veranderingen zijn metabole alkalose, hypocalciurie en hyperreninemisch hyperaldosteronisme.4 Milde tot matige hypofosfatemie wordt vaak waargenomen.5 Ernstige hypofosfatemie met ernstige hyponatremie werd ook gemeld.6,7
De eerste symptomen van GS treden op bij kinderen of jonge volwassenen met een normale groei en een geschiedenis van zoutverslavend gedrag (kinderen die graag augurken of pekel consumeren, gezouten komkommers, sinaasappels en citroenen, kinderen die zout van chips likken, etc.).8 De klinische presentatie varieert van patiënt tot patiënt. Sommigen zijn asymptomatisch, maar anderen ontwikkelen levensbedreigende complicaties. Mannen vertonen een ernstiger fenotype dan vrouwen.8 De meest voorkomende symptomen zijn spierkrampen en -zwakte, constipatie, nocturie, polyurie, dorst, polydipsie, hartritmestoornissen, paresthesieën en een verhoogde zouthonger. Arteriële hypotensie komt vaak voor en is in veel gevallen het meest prominente symptoom, maar in de ouder wordende GS-populatie kan hypertensie voorkomen.8 De correlatie tussen biochemische afwijkingen en symptomen is niet sterk.9 GS interfereert niet met de stemmingen en sociale relaties van kinderen.9,10 Anderszins komen symptomen vaker voor bij volwassenen en kunnen een negatieve invloed hebben op hun kwaliteit van leven. Vijfenveertig procent van de GS-patiënten beschouwt hun symptomen als een matig tot groot probleem.11 Extreme uitputting, spierzwakte, paresthesieën, ernstige vermoeidheid en hypotensie gaan gepaard met een milde tot ernstige vermindering van de dagelijkse activiteiten.9
Geschatte prevalentie van GS is 1:40.0008 en de prevalentie van heterozygotie is ten minste 1% in de Europese bevolking. Tot nu toe zijn meer dan 180 verschillende mutaties in SLC12A3 beschreven.12
Case report
Een 26-jarige man werd in het ziekenhuis opgenomen wegens het optreden van syncope, gegeneraliseerde en gespierde zwakte en spierkrampen. Uit de voorgeschiedenis van de patiënt bleek een episode van syncope met kaliumgehalte 3,16mmol/l. Bij verdere follow-up in de poliklinische evaluatie, werd recurrente incidentie van hypokaliëmie (de laagste waarde 2,6mmol/l) waargenomen. Bloeddruk was normaal 110/80, hartslag was 72 per minuut; er was geen verandering in lichamelijk onderzoek. Geen neurologische bevindingen, gewicht 74kg, lengte 178cm. Bij opname in het ziekenhuis toonde laboratoriumonderzoek milde hypokaliëmie (K+ 3,0mmol/l), hypomagnesiëmie (Mg2+ 1,36mg/dl), hypocalciurie (
mg/24h), en metabole alkalose (HCO3- 29,7mmol/l, BE 5,3mmol/l). De nierfunctie was goed met eGFR>60ml/min. Beeldvormingsonderzoeken waren onopvallend, evenals het ECG.
Verder onderzoek bevestigde dat hypokaliëmie werd veroorzaakt door renale kaliumverspilling. De 24-uurs kaliumverspilling, transtubulaire K+ gradiënt (TTKG), fractionele K+ excretie (FeK) en willekeurige K/creatinine ratio (K/Cr) waren typerend voor hyperkaliurie en staan vermeld in tabel 1.13-15 De dagelijkse magnesiurie bedroeg 64mg/24u, met een fractionele magnesium (FeMg)-uitscheiding van 15%.
De waarden die diagnostisch zijn voor hypokaliëmie.13-15
| Values diagnostic for inappropriate kaliuresis | Patient | |
|---|---|---|
| Urinary K+ | >20mmol/24h Als serum K+3.5mmol/l |
57mmol/l (serum K+=3.0mmol/l) |
| FeK=×100% | >6,5% Als serum K+3.5mmol/l |
12% (serum K+=3,0mmol/l) |
| TTKG= | TTKG>2 Als serum K+3.5mmol/l |
7,3 (serum K+=3,0mmol/l) |
| uK/Cr – willekeurige urine K+ tot creatinine ratio | uK/Cr>15mmol/g Als serum K+3.5mmol/l |
67mmol/g (serum K+=3.0mmol/l) |
Hoewel de bloeddruk normaal was, werden hormonale testen om de ziekte van Conn uit te sluiten en de abdominale CT uitgevoerd, en deze brachten geen afwijkingen in de bijnieren aan het licht. Secundair hyperaldosteronisme met niveaus van aldosteron 289pg/ml (normaal bereik 20-180) en renine 205mIU/ml (normaal bereik 2,8-39,9) waren typerend voor GS.
De patiënt werd gediagnosticeerd als GS op basis van een klinisch fenotype. Na de diagnose werd behandeling met supplementen van kalium en magnesium ingevoerd. Hij kreeg 20mmol kaliumchloride en 18mmol magnesiumpyrrolidoncarboxylaat, met klinische verbetering. Uit de familiegeschiedenis bleek een milde asymptomatische hypokaliëmie bij de 35-jarige zus van de patiënt. Maar onderzoek toonde noch hyperkaliurie noch hypomagnesemie aan. GS werd in dit geval niet gediagnosticeerd.
Genetische analyse
Volgens de genetische analyse, algoritme voorgesteld door Nozu16SLC12A3 gen moet worden getest bij patiënten met hypokalemische metabole alkalose, met voldragen geboorte, normaal gewicht zonder nefrocalcinose, met hypocalcurie en hypomagnesemie.
In het beschreven geval werden genetische tests uitgevoerd en twee heterozygote mutaties: c.35_36insA en c.1095+5G>A werden gevonden in transcript NM_000339.2 van het SLC12A3 gen (Fig. 1). Beide mutaties zijn nog niet in de HGMD(r) database versie beschikbaar . De eerste mutatie werd ook gevonden bij de moeder van de patient en de tweede bij de vader. Slechts een van de twee mutaties die bij onze patiënt werden vastgesteld c.35_36insA werd bij zijn zuster gevonden.
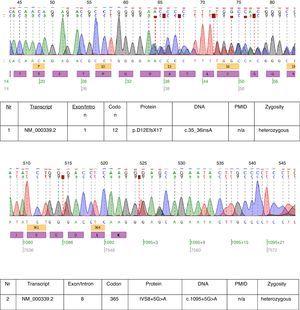
Chromatogrammen van de DNA-sequencing van de getroffen patiënt.
De gevonden mutaties bij deze patiënt bewijzen bijna de klinische diagnose van GS. De pathogenetische relevantie van de frameshift mutatie is duidelijk. Over de pathogenetische relevantie van de splice site mutatie van intron 8 kunnen we niet met zekerheid zeggen. De consensus splice sequentie G (100%), T (100%), A (62%), A (68%), G (82%), T (63%) bij de splice donor van intron 8 komt niet exact overeen met de consensus GTgAGc. De bij deze patiënt gevonden mutatie wijkt de sequentie verder af van de consensus (GtgAac). Ook in silico analyse door mutation taster wordt deze mutatie als ziekteveroorzakend beschouwd.17
Discussie
Tubulopathieën zijn zeldzame ziekten. Volgens de RenalTube database18 zijn de meest voorkomende primaire tubulopathieën in de Europese bevolking distale renale tubulaire acidose, Bartter syndroom, familiaire hypomagnesiëmie met hypercalciurie en GS.18 De prevalentie van GS is ongeveer 25 gevallen per 1 miljoen.3 Omdat GS een van de meest voorkomende is, zullen waarschijnlijk veel nefrologen en endocrinologen tijdens hun loopbaan geconfronteerd worden met gevallen van GS. Onze patiënt had een typische klinische presentatie van GS en reageerde goed op de therapie. Het probleem met GS is dat over het hoofd geziene hypokaliëmie de dood kan veroorzaken door hartstilstand of verlamming van de ademhalingsspieren. Het ernstige neuromusculaire symptoom zoals hypokalemische verlamming komt voor bij maximaal 6% van de patiënten (meer voor bij Aziatische patiënten).3
De erfelijke tubulopathieën hebben een soort “spiegelbeelden”-verworven tubulopathieën veroorzaakt door diuretica. Elk diureticum (om precies te zijn: natriuretica, acquaretica of glucuretica) veroorzaken afwijkingen die lijken op die welke in erfelijke tubulopathieën worden gevonden (tabel 2). Bijna in elke tubulopathie treedt polyurie op, en veranderingen in de kaliumspiegel behoren tot de meest voorkomende problemen. Bij patiënten met GS worden mutaties van het SLC12A3-gen gevonden. Dit gen codeert voor de thiazide-gevoelige transporter. Daarom lijkt GS op chronische behandeling met thiaziden.
Diuretica en tubulopathieën.
| Erfelijk enkelvoudig gendefect | Potassiumconcentratie | Symptomen vergelijkbaar met gebruik van: | Gene | Gene product | Gene product locatie |
|---|---|---|---|---|---|
| Guibaud-Vainsel-syndroom (RTA 3) | ↓ | Acetazolamide | CA2 | CA II | Proximale tubulus |
| Renale glycosurie | – | SGLT2-remmersa (Canagliflozin, Tofogliflozin, Dapagliflozin) | SLC5A2 | Sodium/glucose cotransporter 2 (SGLT2) | Proximale tubulus |
| Bartter-syndroom | ↓ | Furosemide | SLC12A1b | Na-K-2Cl cotransporter (NKCC2) | Loop van Henle |
| Gitelman syndroom | ↓ | Thiazide | SLC12A3 | Thiazide-gevoelige Na-Cl cotransporter | Distal tubule |
| Diabetes insipidus | – | Vaptan | AVPR2, AQP2 | V2 receptor | Collecting duct |
| Pseudohypoaldosteronizm, AD | ← | Spironolacton | NR3C2 | Spironolacton-gevoelige mineralocorticoïdreceptor | Collecting duct |
| Pseudohypoaldosteronizm, AR | Amiloride | SCNN1A, SCNN1B, SCNN1G | Amiloride-gevoelig epitheliaal natriumkanaal | Collecting duct | |
AR – autosomaal recessief; AD – autosomaal dominant.
SGLT2-remmers (glucuretica) zijn de nieuwe geneesmiddelen voor de behandeling van type 2-diabetes; tijdens de behandeling worden osmotische diurese-gerelateerde gebeurtenissen zoals pollakiurie en polyurie waargenomen.
Hoewel er verschillende Bartter-genen zijn, is dit het gen dat furosemid nabootst.
Aanpak hypokaliëmie
GS wordt ook wel familiaire hypokaliëmische hypomagnesiëmie genoemd, omdat hypokaliëmie het meest voorkomende verschijnsel is. Bij een patiënt met hypokaliëmie en verdenking op tubulopathie dient men hyperkaliurie te bevestigen. Renale kaliumverspilling kan worden aangetoond door berekening van TKKG, FeK, willekeurige K/Cr-ratio of na 24-uurs urineverzameling (tabel 1). Kaliurie moet worden vastgesteld wanneer de patiënt geen diuretica of kaliumsupplementen gebruikt en wanneer de kaliumspiegel laag is. Wanneer de kaliumexcretie lager is dan 30 mmol/l en de TTKG laag is, wordt de hypokaliëmie veroorzaakt door extrarenaal verlies of een transcellulaire K+ shift.
Wanneer overmatig kaliumverlies door de nieren bij een patiënt met hypokaliëmie wordt bevestigd, wordt de verdere diagnose gebaseerd op pH en bloeddruk. In veel gevallen zullen deze eenvoudige onderzoeken voldoende zijn voor de diagnose. Een belangrijk probleem dat steeds meer belangstelling krijgt is primair aldosteronisme (PA). Sommige klinische studies hebben gesuggereerd dat PA de oorzaak is van meer dan 10% van de arteriële hypertensie (AH), en vaker voorkomt bij patiënten met AH die resistent zijn tegen antihypertensiva.19 Hypokaliëmie is een van de “klassieke” symptomen van PA, maar komt niet zo vaak voor als werd gedacht.20 Toch is het, omdat er geen gouden standaard is voor de diagnose van PA, de moeite waard om te denken aan de RAA-as bij patiënten met AH en hypokaliëmie.19
Diagnose van hypomagnesiëmie
Hypomagnesiëmie is een veel voorkomende afwijking bij GS-patiënten, hoewel deze niet in alle gevallen wordt waargenomen.21 Bij normomagnesiënemische patiënten met GS zijn de klinische manifestatie en de elektrolytafwijkingen milder.22
Gelijk aan kaliumionen worden magnesiumionen vrij gefilterd door de glomeruli. 10% van het gefiltreerde Mg2+ wordt geabsorbeerd door de proximale tubulus, 50-70% in de opgaande ledemaat van de lus van Henle. Distale reabsorptie is afhankelijk van epitheliale Mg2+ TRPM6 kanalen. Magnesiurie is afhankelijk van orale inname en FeMg moet worden berekend om renale Mg2+ verspilling vast te stellen (FeMg=(urine magnesium×serum creatinine)/×100). Een FeMg van minder dan 2% wijst op een slechte inname, verlies via de darm of verplaatsing van Mg naar de cellen. Een FeMg van meer dan 4% wijst op renale Mg2+-verspilling. Als renale Mg-verspilling is aangetoond, moet de willekeurige Ca/Cr-verhouding worden berekend. Urine Ca/Cr-ratio 0,3 is typisch voor hypermagesurie bij het Bartter-syndroom, evenals geïsoleerde recessieve hypomagnesiëmie met normocalciurie, familiaire hypomagnesiëmie met hypercalciurie en nefrocalcinose, autosomaal dominante hypocalciëmie met hypercalciurie, behandeling met lusdiuretica en nefropathie veroorzaakt door sommige nefrotoxinen.23
Magnesium- en kaliumhomeostase zijn aan elkaar gerelateerd, en kaliumdepletie kan pas gecorrigeerd worden na correctie van hypomagnesiëmie.23 Gemiddeld dienen patiënten tot 500mEq kalium, 4-5mg/kg/dag 5-10mg magnesiumchloride te krijgen. Amiloride (5-10mg/dag) en spironolacton (200-300mg) zijn nuttig.3
Clinische symptomen en laboratoriumuitslagen zijn essentieel voor de diagnose. In gevallen met een typisch fenotype bevelen sommige auteurs het uitvoeren van een thiazidetest aan om de diagnose te bevestigen.24 In het gepresenteerde geval bracht genetisch onderzoek twee nieuwe mutaties van het SLC12A3-gen aan het licht. Ze worden waarschijnlijk veroorzaakt door het ontbreken van functionele mutaties van NCCT.
Conclusies
GS syndroom is een van de zeldzame oorzaken van hypokaliëmie, en het lijkt een uitdaging voor artsen. Wij tonen in dit artikel aan dat als men zich een zeer eenvoudige aanpak van hypokaliëmie herinnert en zich bewust is van de werking van diuretica en hun gelijkenis met erfelijke tubulopathieën, de diagnose vrij ongecompliceerd kan zijn. GS moeten worden onderscheiden van andere tubulopathieën (zowel erfelijke als verworven) en andere oorzaken van hypokaliëmie (bv. de ziekte van Conn). Familiaire voorgeschiedenis kan asymptomatische patiënten met GS aan het licht brengen. Geschikte behandeling beschermt patiënten tegen potentieel gevaarlijke complicaties.