
Een korte inleiding tot ChIP-Seq

Proteïne-DNA-interacties worden veel gebruikt om de mechanismen die aan de celfysiologie ten grondslag liggen te ontrafelen. De ontwikkeling van de chromatine-immunoprecipitatietechnologie (ChIP) heeft de bestudering van dergelijke mechanismen mogelijk gemaakt. Na verdere ontwikkeling ontstond een deep sequencing alternatief (ChiP-Seq), dat voordelen biedt in termen van specificiteit en gevoeligheid.
Een ChIP-Seq experiment begint met een cross-link van de hele cel met formaldehyde, gevolgd door sonicatie en DNA-isolatie. Daarna wordt immunoprecipitatie van het DNA-eiwitcomplex uitgevoerd, dat bestaat uit antilichamen die zich binden aan specifieke eiwitten. De gevormde immuno-complexen worden geprecipiteerd en gezuiverd. Ten slotte wordt het DNA gesequeneerd, waardoor gegevens met een hoge resolutie van verrijkte plaatsen worden verkregen. Deze aanpak, samen met een gevestigde ChIP-seq pijplijn, stelt onderzoekers in staat om DNA-transcriptiefactoren, histon-modificatie sites, epigenetische veranderingen, en gen-regelgeving netwerk handtekeningen vast te leggen.
Clinische relevantie en toepassingen
Epigenetische onevenwichtigheden in ziekte en gezondheid aandoeningen kunnen histon-modificatie en veranderde transcriptiefactoren te betrekken. In dit verband zijn ChIP-Seq studies gebruikt om pathologische moleculaire mechanismen op te helderen die ten grondslag liggen aan kanker en andere ziekten. ChIP-seq analyse draagt ook bij tot het begrijpen van de rol van transcriptiefactoren tijdens ziekten. In feite lijken sommige transcripten te worden gewijzigd tijdens klinische fenotype manifestaties.
Overzicht van de ChIP-Seq pijplijn
De ChIP-Seq analyse pijplijn is de belangrijkste component van DNA-eiwit interactie projecten en bestaat uit verschillende stappen, waaronder ruwe data verwerking, kwaliteitscontrole analyse, uitlijning aan het referentiegenoom, kwaliteitscontrole van de uitgelijnde leest, peak calling, annotatie, en visualisatie. Een doordachte experimentele opzet is echter cruciaal voor het verkrijgen van resultaten van hoge kwaliteit in een ChIP-seq experiment. Voordat u met uw analyse begint, is het essentieel om parameters zoals monsterreplicaten, controlegroepen, sequencing-kits en sequencing-platforms te overwegen.
Kwaliteitscontrole
Alle Basepair-rapporten bieden kwaliteitsscores om te helpen bij het blootleggen van potentiële sequencing-problemen of verontreiniging in uw inputdata.
De stap Kwaliteitscontrole (QC) is gericht op het evalueren van de kwaliteit van high throughput-gegevens die zijn gegenereerd door sequencing. Deze stap is vergelijkbaar met die welke wordt uitgevoerd in DNA-seq en RNA-seq analyses. Hier omvatten de belangrijkste geëvalueerde metrieken sequentie- en base-kwaliteit, GC-gehalte, aanwezigheid van sequencing-adapters en oververtegenwoordigde sequenties. Een van de meest gebruikte programma’s voor dit soort analyse is FastQC. Indien sequenties van lage kwaliteit worden geïdentificeerd, kunnen deze later worden verwijderd tijdens de trimstap. Hoewel het een optionele stap, trimmen verbetert de kwaliteit van de gegevens door het behoud van alleen van hoge kwaliteit reads.
Alignment
Na QC meting, ChIP-Seq gelezen worden uitgelijnd met een referentie-genoom. Read mapping stelt onderzoekers in staat om de oorsprong van een gelezen sequentie in het genoom te identificeren. Populaire uitlijningstools zijn Bowtie en BWA, die beide worden gebruikt in de ChIP-seq pijplijnen van Basepair. Beide tools brengen laag-divergente sequenties in kaart tegen een referentie-genoom.
De read count flow helpt een groot-picture overzicht van bruikbare leest aan het einde van het trimmen, uitlijnen, en deduplicatie processen. Zie de figuur als een data-analyse assemblagelijn: input ruwe gegevens, krijg een output van bruikbare leest.
Kwaliteitscontrole van de uitgelijnde leest
De volgende stap bestaat uit QC inferentie van de uitgelijnde dataset. Tijdens het mapping proces, kunnen lees duplicaten geïntroduceerd door PCR amplificatie en sequencing biases veroorzaken tijdens peak calling en verrijkings analyse. Basepair gebruikt het Picard gereedschap om duplicaten te verwijderen. Zodra duplicaten zijn verwijderd, moet u de Non-Redundant Fraction (NRF) van de uitgelijnde leest evalueren. NRF meet de unieke gelezen mapping met het referentie genoom. Ideale ChIP-seq experimenten moeten minder dan drie reads per positie hebben.
Peak Calling
De peak calling stap detecteert verrijkte eiwit-DNA interactie regio’s op het genoom. Basepair’s ChIP-seq pijplijn gebruikt MACS2 om deze analyse uit te voeren. In MACS2 wordt peak calling uitgevoerd op basis van drie hoofdstappen: fragmentschatting, gevolgd door identificatie van lokale ruisparameters en dan piekidentificatie. Als output van deze stap krijgen de gebruikers een definitieve tabel met piekinformatie, zoals verrijkingscore, -log10pwaarde, -log10qwaarde en de positie aan het begin van de piek. Het gebruik van controlemonsters wordt in deze stap sterk aanbevolen voor vergelijking met de onderzochte doeldataset. Bedenk dat goede controlegroepen betrouwbaardere resultaten opleveren.
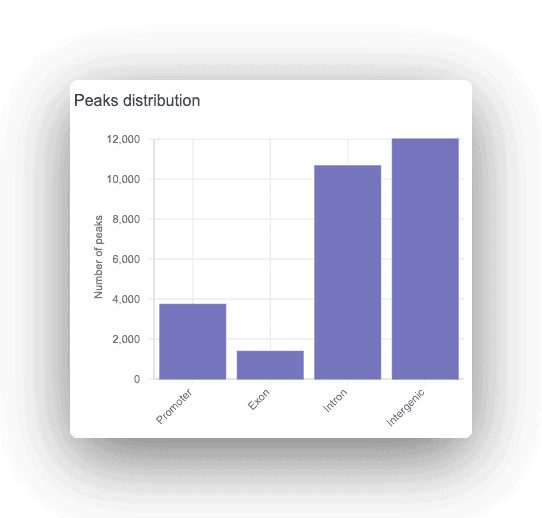
Elke piek wordt geannoteerd als promoter, intronic, of intergenic, waarbij het corresponderende gen wordt weergegeven. Voor alle gevonden pieken, wordt een motief analyse gedaan om oververtegenwoordigde transcriptiefactor bindingsplaatsen te vinden.
Overzicht van de resultaten
Een ChIP-seq pijplijn kan niet alleen informatie over de chromatine staat, maar ook transcriptiefactor binding in een bepaald gen of loci context. Het voorkomen van histon-modificaties en transcriptiefactoren in DNA-regulerende regio’s kan een toestand-specifieke epigenetische handtekening vormen. Zo kunnen epigenetische verstoringen geassocieerd worden met klinische fenotypes. Zo kan heterogeniteit van chromatinetoestanden bijvoorbeeld leiden tot resistentie tegen behandeling bij borstkanker. Deze cellen hebben de neiging om repressieve histon-modificaties markers verliezen en verder verhogen van de expressie van genen waarvan bekend is dat weerstand tegen behandeling van kanker te bevorderen.
Peak, Motif en Pathway Analysis in ChIP-Seq Analysis Pipeline
De identificatie van motief transcriptiefactor verrijking wordt gebruikt om te verduidelijken of transcriptiefactoren samenwerken of concurreren in een bepaald gebied. De identificatie van pieken in DNA-motiefgebieden kan de interpretatie van de experimentele resultaten verbeteren. Tezamen geven zowel piek- als motiefanalyses inzicht in wat er in een cel kan gebeuren. De integratie van piek- en motiefverrijkingen resulteert in een epigenomisch landschap met mogelijke biologische gevolgen. Verder wordt pathway-analyse gebruikt om eiwitten in een pathway te identificeren. Onderzoeken en conclusies worden geformuleerd op basis van de aanwezigheid van een eiwit.
Datavisualisatie
Resultaatgegevens van een ChIP-seq pipeline kunnen worden gevisualiseerd met behulp van een genoom-browser. Basepair rapporten bevatten een ingebedde IGV2 genoom-browser die u toelaat om te interageren met uw gegevens. De gegevens kunnen ook worden gevisualiseerd met behulp van heatmaps, representatieve infographics op basis van gegevensdichtheid die de aan- of afwezigheid van specifieke markeringen laten zien. Andere grafieken die hier worden gebruikt zijn enrichment plot, upSet en coverage plot, waarmee zowel de dekking van piekregio’s over het genoom wordt berekend als weergegeven.
De genome browser is een geweldig hulpmiddel voor het visualiseren van uw ruwe genomische gegevens. Het is ingebouwd in elk ChIP-seq analyse rapport op Basepair.
1. Grosselin, K., A. Durand, et al. High-throughput single-cell ChIP-seq identificeert heterogeniteit van chromatine toestanden in borstkanker. Nat Genet, v.51, n.6, Jun, p.1060-1066. 2019.
2. Northrup, D. L. e K. Zhao. Toepassing van ChIP-Seq en verwante technieken voor de studie van immuunfunctie. Immunity, v.34, n.6, 24 jun, p.830-42. 2011.
3. Park, S. J., J. H. Kim, et al. A ChIP-Seq Data Analysis Pipeline Based on Bioconductor Packages. Genomics Inform, v.15, n.1, mrt, p.11-18. 2017.
4. Pepke, S., B. Wold, et al. Computation for ChIP-seq and RNA-seq studies. Nat Methods, v.6, n.11 Suppl, Nov, p.S22-32. 2009.
5. Satoh, J., N. Kawana, et al. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul Syst Bio, v.7, p.139-52. 2013.