
Vnitřní transkribované mezery (ITS) jsou oblastí v ribozomálním transkriptu, která je během zrání vyříznuta a degradována. Jejich sekvence obecně vykazují větší variabilitu než ribozomální sekvence, díky čemuž jsou oblíbené pro fylogenetickou analýzu a/nebo identifikaci druhů a kmenů. Zejména u hub je to oblíbená identifikační technika, protože identifikace na základě morfologických znaků je pracná a často nevede ke správnému výsledku. Použitá oblast je obvykle kombinací ITS a ribozomálních sekvencí a nejčastěji se skládá z (částečné) sekvence 18S rRNA (16S rRNA u prokaryot), vnitřně přepisované oblasti (ITS1), celé sekvence 5,8s rRNA, vnitřně přepisované oblasti (ITS2) a (částečné) sekvence 28s rRNA. Mnoho sekvencí ITS je k dispozici ve veřejných databázích, jako jsou NCBI a EBI.
Pracovní postup typizace ITS v softwaru BIONUMERICS
V softwaru BIONUMERICS lze sekvence ITS dávkově importovat ze stopových souborů pomocí zásuvného modulu pro dávkové sestavování sekvencí. Referenční sekvence lze importovat z veřejných databází a/nebo ze souborů fasta nebo GenBank.
K vícenásobnému zarovnání sekvencí lze použít několik algoritmů: Needleman-Wunsch, Wilbur-Lipman a náš vlastní algoritmus vypracovaný v Applied Maths. Zarovnání je možné upravit i ručně. Na základě vícenásobného zarovnání lze vypočítat shlukování, které odráží fylogenetické vztahy mezi analyzovanými sekvencemi.
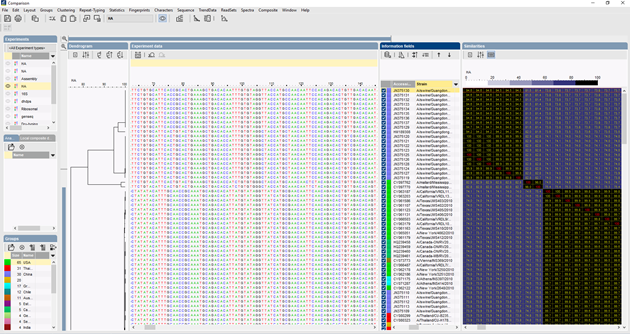
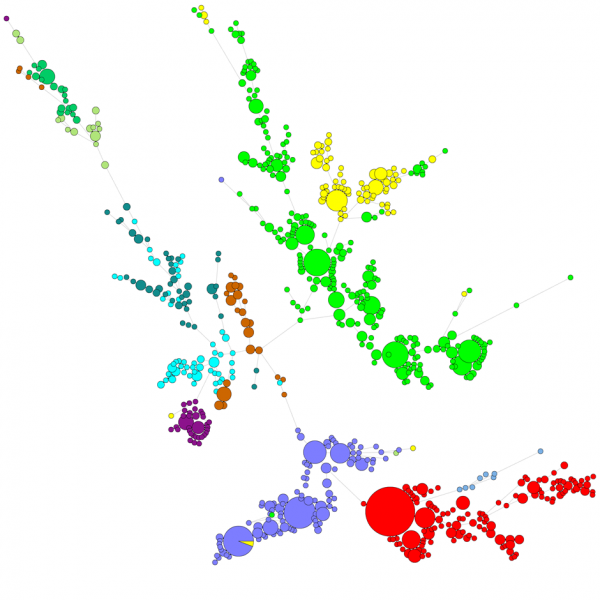
Při porovnávání velkého počtu sekvencí neposkytuje dendrogram UPGMA nebo sousedního spojování příliš dobrý přehled o datech. V těchto případech poskytuje lepší řešení nezakořeněný strom, v programu BIONUMERICS je možné z dat sekvencí vypočítat stromy minimálního rozpětí, stromy maximální věrohodnosti a stromy maximální parsimonie. Pro data lze definovat různé skupiny a tyto skupiny jsou vizualizovány ve stromech, což poskytuje pěkný přehled o fylogenetických vztazích, shlucích přítomných v databázi a také odlehlých hodnotách. Na obrázku níže je znázorněn minimální rozpínací strom založený na sekvencích ITS, skupiny představují různé rody.
Sekvence ITS lze také použít k identifikaci neznámých organismů oproti databázi. Pomocí modulu Classifiers and Identification (Klasifikátory a identifikace) může uživatel vytvořit identifikační projekty obsahující identifikované kmeny a použít je k identifikaci neznámého kmene; na základě podobnosti bude přiřazeno skóre spolu se spolehlivostí tohoto skóre. Uživatel má plnou kontrolu nad parametry pro porovnávání a mezními hodnotami pro identifikaci.