
Les espaceurs transcriptionnels internes (ITS) sont des régions au sein du transcrit ribosomal qui sont excisées et dégradées pendant la maturation. Leurs séquences présentent généralement plus de variations que la séquence ribosomique, ce qui les rend populaires pour l’analyse phylogénétique et/ou l’identification des espèces et des souches. En particulier pour les champignons, il s’agit d’une technique d’identification populaire car l’identification basée sur les caractéristiques morphologiques est laborieuse et ne conduit souvent pas à un résultat correct. La région utilisée est généralement une combinaison de séquences ITS et ribosomiques et se compose le plus souvent d’une séquence (partielle) d’ARNr 18S (ARNr 16S pour les procaryotes), d’une région transcrite en interne (ITS1), de la séquence entière d’ARNr 5.8s, d’une région transcrite en interne (ITS2) et d’une séquence (partielle) d’ARNr 28s. De nombreuses séquences ITS sont disponibles sur des bases de données publiques telles que NCBI et EBI.
Flux de travail de typage ITS dans le logiciel BIONUMERICS
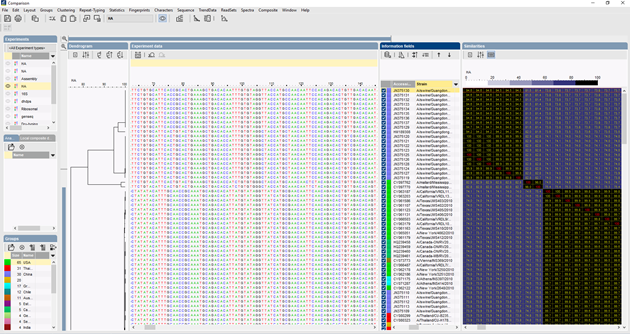
Dans BIONUMERICS, les séquences ITS peuvent être importées en lot à partir de fichiers de traces en utilisant le plugin d’assemblage de séquences en lot. Les séquences de référence peuvent être importées à partir de bases de données publiques et/ou de fichiers fasta ou GenBank.
Plusieurs algorithmes peuvent être utilisés pour effectuer un alignement multiple des séquences, Needleman-Wunsch, Wilbur-Lipman et notre propre algorithme propriétaire élaboré à Applied Maths. Une adaptation manuelle de l’alignement est possible. Sur la base de l’alignement multiple, un clustering peut être calculé pour refléter les relations phylogénétiques entre les séquences analysées.
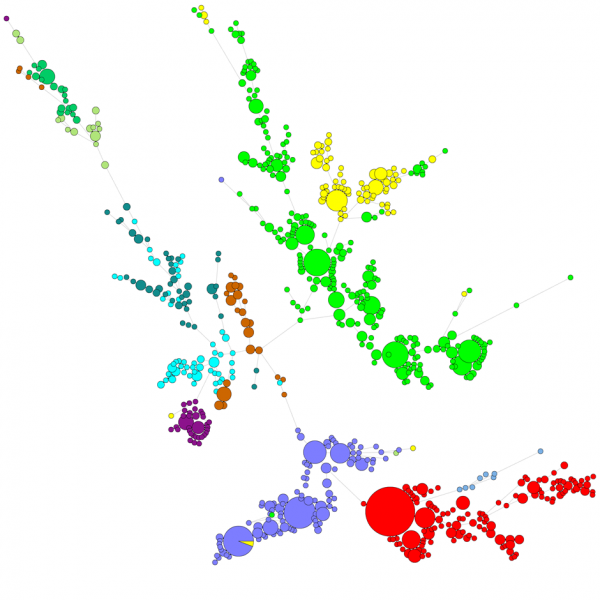
Lorsque l’on compare un grand nombre de séquences, un dendrogramme UPGMA ou à jonction de voisins ne donne pas une très bonne vue d’ensemble des données. Dans ces cas, un arbre non raciné fournit une meilleure solution. Dans BIONUMERICS, il est possible de calculer des arbres à portée minimale, des arbres de vraisemblance maximale et des arbres de parcimonie maximale à partir de données de séquences. Différents groupes peuvent être définis pour les données et ces groupes sont visualisés dans les arbres, ce qui donne une bonne vue d’ensemble des relations phylogénétiques, des groupes présents dans la base de données et également des valeurs aberrantes. La figure ci-dessous montre un arbre à portée minimale basé sur les séquences ITS, les groupes représentent différents genres.
La séquence ITS peut également être utilisée pour identifier des organismes inconnus par rapport à une base de données. Avec le module Classificateurs et Identification, l’utilisateur peut créer des projets d’identification contenant des souches identifiées et les utiliser pour identifier une souche inconnue, un score sera attribué en fonction de la similarité ainsi que de la fiabilité de ce score. L’utilisateur a un contrôle total sur les paramètres de comparaison et les valeurs seuils pour l’identification.