
Interne transkribierte Spacer (ITS) sind Bereiche innerhalb des ribosomalen Transkripts, die während der Reifung ausgeschnitten und abgebaut werden. Ihre Sequenzen weisen im Allgemeinen eine größere Variation auf als die ribosomale Sequenz, was sie für phylogenetische Analysen und/oder die Identifizierung von Arten und Stämmen interessant macht. Vor allem bei Pilzen ist dies eine beliebte Methode zur Identifizierung, da die Identifizierung anhand morphologischer Merkmale mühsam ist und oft nicht zu einem korrekten Ergebnis führt. Die verwendete Region ist in der Regel eine Kombination aus ITS- und ribosomalen Sequenzen und besteht meist aus einer (partiellen) 18S rRNA-Sequenz (16S rRNA für Prokaryoten), einer intern transkribierten Region (ITS1), der gesamten Sequenz der 5,8s rRNA, einer intern transkribierten Region (ITS2) und einer (partiellen) Sequenz der 28s rRNA. Viele ITS-Sequenzen sind in öffentlichen Datenbanken wie NCBI und EBI verfügbar.
ITS-Typisierungs-Workflow in der BIONUMERICS Software
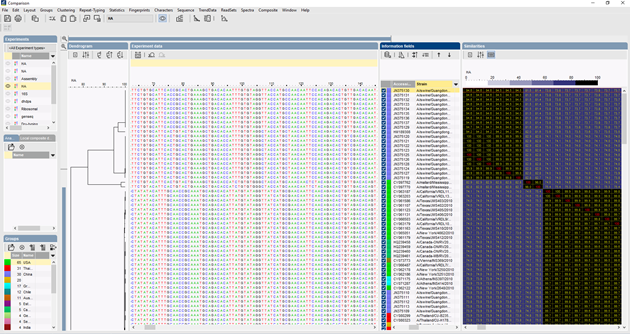
In BIONUMERICS können die ITS-Sequenzen im Batch aus Trace-Dateien mit dem Batch-Sequenzassembly-Plugin importiert werden. Referenzsequenzen können aus öffentlichen Datenbanken und/oder aus fasta- oder GenBank-Dateien importiert werden.
Es können verschiedene Algorithmen verwendet werden, um ein multiples Alignment der Sequenzen durchzuführen: Needleman-Wunsch, Wilbur-Lipman und unser eigener Algorithmus, der bei Applied Maths entwickelt wurde. Eine manuelle Anpassung des Alignments ist möglich. Auf der Grundlage des multiplen Alignments kann ein Clustering berechnet werden, das die phylogenetischen Beziehungen zwischen den analysierten Sequenzen widerspiegelt.
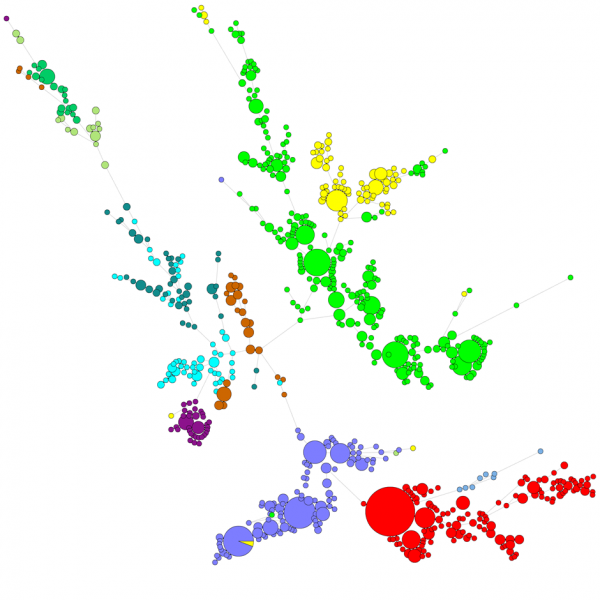
Beim Vergleich einer großen Anzahl von Sequenzen gibt ein UPGMA- oder Nachbarschafts-Dendrogramm keinen sehr guten Überblick über die Daten. In diesen Fällen bietet ein nicht verwurzelter Baum eine bessere Lösung. In BIONUMERICS ist es möglich, ausgehend von Sequenzdaten minimal spanning trees, maximum likelihood trees und maximum parsimony trees zu berechnen. Verschiedene Gruppen können für die Daten definiert werden und diese Gruppen werden in den Bäumen visualisiert, was einen schönen Überblick über die phylogenetischen Beziehungen, die in der Datenbank vorhandenen Cluster und auch die Ausreißer gibt. Die Abbildung unten zeigt einen minimal spannenden Baum auf der Grundlage von ITS-Sequenzen, wobei die Gruppen verschiedene Gattungen darstellen.
Die ITS-Sequenz kann auch verwendet werden, um unbekannte Organismen anhand einer Datenbank zu identifizieren. Mit dem Klassifizierungs- und Identifizierungsmodul kann der Benutzer Identifizierungsprojekte erstellen, die identifizierte Stämme enthalten, und diese verwenden, um einen unbekannten Stamm zu identifizieren. Der Benutzer hat die volle Kontrolle über die Parameter für den Vergleich und die Grenzwerte für die Identifizierung.