
A belső átírt szóközök (ITS) a riboszómális transzkriptumon belüli régió, amely az érés során kivágódik és lebomlik. Szekvenciáik általában több variációt mutatnak, mint a riboszómális szekvencia, ezért népszerűek a filogenetikai elemzéshez és/vagy a fajok és törzsek azonosításához. Különösen a gombák esetében ez egy népszerű azonosítási technika, mivel a morfológiai jellemzők alapján történő azonosítás munkaigényes és gyakran nem vezet helyes eredményre. A használt régió általában az ITS és a riboszómális szekvenciák kombinációja, és leggyakrabban a 18S rRNS (prokarióták esetében a 16S rRNS) (részleges) szekvenciájából, egy belső átírt régióból (ITS1), az 5,8s rRNS teljes szekvenciájából, egy belső átírt régióból (ITS2) és a 28s rRNS (részleges) szekvenciájából áll. Számos ITS-szekvencia elérhető a nyilvános adatbázisokban, mint például az NCBI és az EBI.
ITS tipizálási munkafolyamat a BIONUMERICS szoftverben
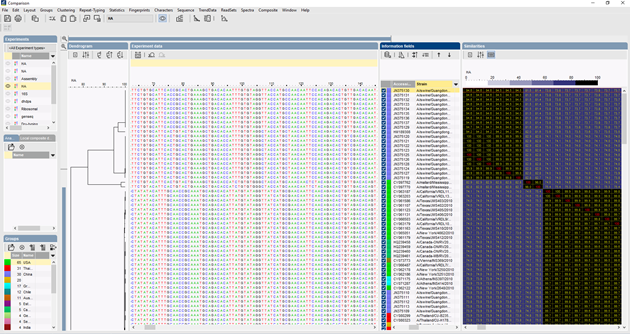
A BIONUMERICS-ben az ITS-szekvenciákat a Batch szekvencia-összeszerelő plugin segítségével tételesen lehet importálni a nyomkövetési fájlokból. A referenciaszekvenciák nyilvános adatbázisokból és/vagy fasta- vagy GenBank-fájlokból importálhatók.
A szekvenciák többszörös összehangolásához több algoritmus is használható: Needleman-Wunsch, Wilbur-Lipman és az Applied Maths-nál kidolgozott saját algoritmusunk. Az összehangolás kézi adaptálása is lehetséges. A többszörös igazítás alapján kiszámítható egy klaszterezés, amely tükrözi az elemzett szekvenciák közötti filogenetikai kapcsolatokat.
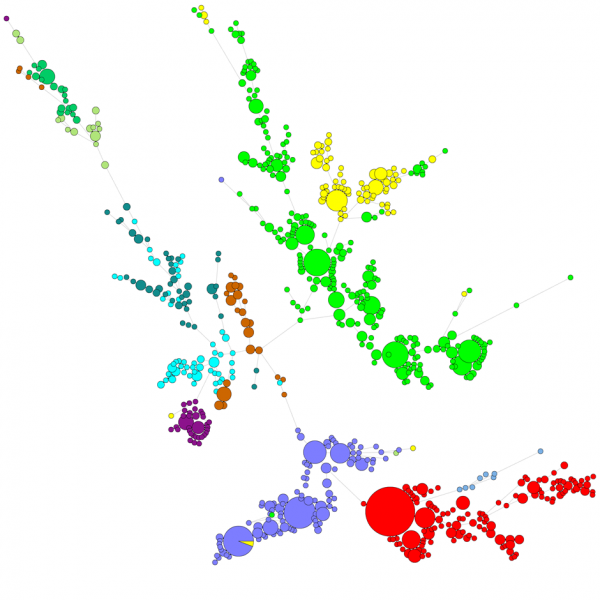
Nagyszámú szekvencia összehasonlításakor az UPGMA vagy a szomszéd-összekötő dendrogram nem ad túl jó áttekintést az adatokról. Ezekben az esetekben egy gyökértelen fa jobb megoldást nyújt, a BIONUMERICS-ben lehetőség van a szekvenciaadatokból kiindulva minimális átfutófák, maximális valószínűségű fák és maximális parszimónia fák kiszámítására. Az adatokhoz különböző csoportokat lehet definiálni, és ezeket a csoportokat a fák vizualizálják, így szép áttekintést adnak a filogenetikai kapcsolatokról, az adatbázisban jelen lévő klaszterekről és a kiugró értékekről is. Az alábbi ábra egy ITS-szekvenciákon alapuló minimális átfedési fát mutat, a csoportok különböző nemzetségeket képviselnek.
Az ITS-szekvencia ismeretlen szervezetek azonosítására is használható egy adatbázis alapján. Az osztályozók és azonosítás modul segítségével a felhasználó azonosított törzseket tartalmazó azonosítási projekteket hozhat létre, és ezeket felhasználhatja egy ismeretlen törzs azonosítására, a hasonlóság alapján pontszámot kap a rendszer a pontszám megbízhatóságával együtt. A felhasználó teljes mértékben kontrollálhatja az összehasonlítás paramétereit és az azonosításhoz szükséges határértékeket.