
Wewnętrzne odstępy transkrybowane (ITS) to regiony transkryptu rybosomalnego, które są wycinane i degradowane podczas dojrzewania. Ich sekwencje wykazują zwykle większą zmienność niż sekwencja rybosomalna, co czyni je popularnymi w analizie filogenetycznej i/lub identyfikacji gatunków i szczepów. Szczególnie w przypadku grzybów jest to popularna technika identyfikacji, gdyż identyfikacja na podstawie cech morfologicznych jest pracochłonna i często nie prowadzi do poprawnego wyniku. Wykorzystywany region jest zwykle kombinacją sekwencji ITS i rybosomalnych i najczęściej składa się z (częściowej) sekwencji 18S rRNA (16S rRNA dla prokariotów), wewnętrznie transkrybowanego regionu (ITS1), całej sekwencji 5,8s rRNA, wewnętrznie transkrybowanego regionu (ITS2) i (częściowej) sekwencji 28s rRNA. Wiele sekwencji ITS jest dostępnych w publicznych bazach danych, takich jak NCBI i EBI.
Praca nad typowaniem ITS w oprogramowaniu BIONUMERICS
W BIONUMERICS, sekwencje ITS mogą być importowane wsadowo z plików śladowych przy użyciu wtyczki batch sequence assembly. Sekwencje referencyjne mogą być importowane z publicznych baz danych i/lub z plików fasta lub GenBank.
Do wykonania wielokrotnego wyrównania sekwencji można użyć kilku algorytmów, Needleman-Wunsch, Wilbur-Lipman oraz naszego własnego algorytmu opracowanego w Applied Maths. Możliwa jest ręczna adaptacja wyrównania. Na podstawie wielokrotnego wyrównania można obliczyć klasteryzację odzwierciedlającą zależności filogenetyczne między analizowanymi sekwencjami.
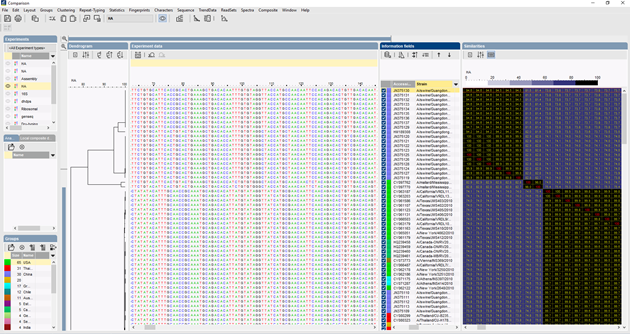
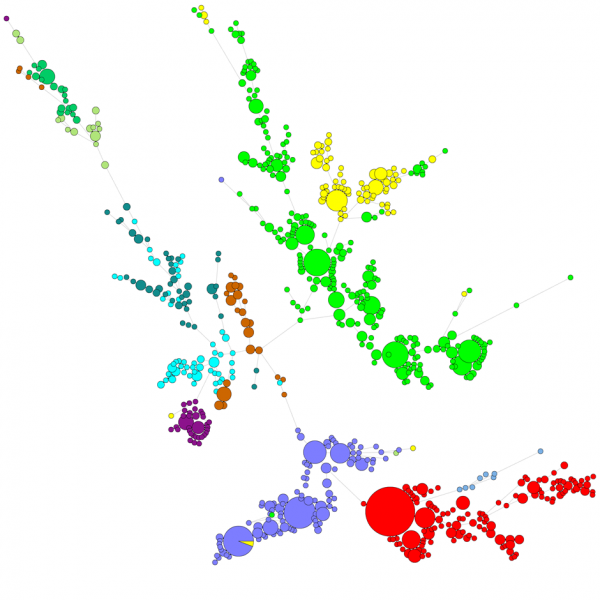
W przypadku porównywania dużej liczby sekwencji, dendrogram UPGMA lub dendrogram sąsiedzki nie daje bardzo dobrego przeglądu danych. W BIONUMERICS możliwe jest obliczenie minimalnych drzew rozpinających, drzew o maksymalnej wiarygodności i drzew o maksymalnej parsymonii na podstawie danych sekwencji. Różne grupy mogą być zdefiniowane dla danych i te grupy są wizualizowane na drzewach, dając ładny przegląd relacji filogenetycznych, klastrów obecnych w bazie danych, a także wartości odstających. Poniższy rysunek przedstawia minimalne drzewo rozpinające oparte na sekwencji ITS, grupy reprezentują różne rodzaje.
Sekwencja ITS może być również użyta do identyfikacji nieznanych organizmów w bazie danych. Dzięki modułowi Classifiers and Identification, użytkownik może tworzyć projekty identyfikacyjne zawierające zidentyfikowane szczepy i używać ich do identyfikacji nieznanego szczepu, wynik zostanie przypisany na podstawie podobieństwa wraz z wiarygodnością tego wyniku. Użytkownik ma pełną kontrolę nad parametrami porównania i wartościami odcięcia dla identyfikacji.