
Los espaciadores transcritos internos (ITS) son regiones dentro del transcrito ribosómico que se extirpan y degradan durante la maduración. Sus secuencias generalmente muestran más variación que la secuencia ribosomal, lo que las hace populares para el análisis filogenético y/o la identificación de especies y cepas. Especialmente en el caso de los hongos, es una técnica popular para la identificación, ya que la identificación basada en las características morfológicas es laboriosa y a menudo no conduce a un resultado correcto. La región utilizada suele ser una combinación de ITS y secuencias ribosómicas y, en la mayoría de los casos, consiste en una secuencia (parcial) del ARNr 18S (ARNr 16S para procariotas), una región transcrita internamente (ITS1), la secuencia completa del ARNr 5,8s, una región transcrita internamente (ITS2) y una secuencia (parcial) del ARNr 28s. Muchas secuencias ITS están disponibles en bases de datos públicas como NCBI y EBI.
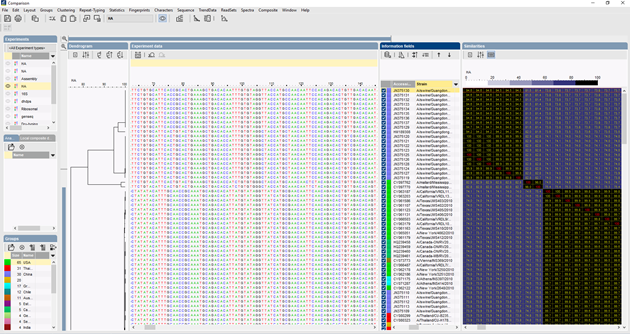
Flujo de trabajo de tipificación de ITS en el software BIONUMERICS
En BIONUMERICS, las secuencias ITS pueden importarse por lotes desde archivos de trazas utilizando el plugin de ensamblaje de secuencias por lotes. Las secuencias de referencia pueden ser importadas de bases de datos públicas y/o de archivos fasta o GenBank.
Se pueden utilizar varios algoritmos para realizar un alineamiento múltiple de las secuencias, Needleman-Wunsch, Wilbur-Lipman y nuestro propio algoritmo elaborado en Applied Maths. Es posible la adaptación manual del alineamiento. Basándose en el alineamiento múltiple, se puede calcular un clustering que refleje las relaciones filogenéticas entre las secuencias analizadas.
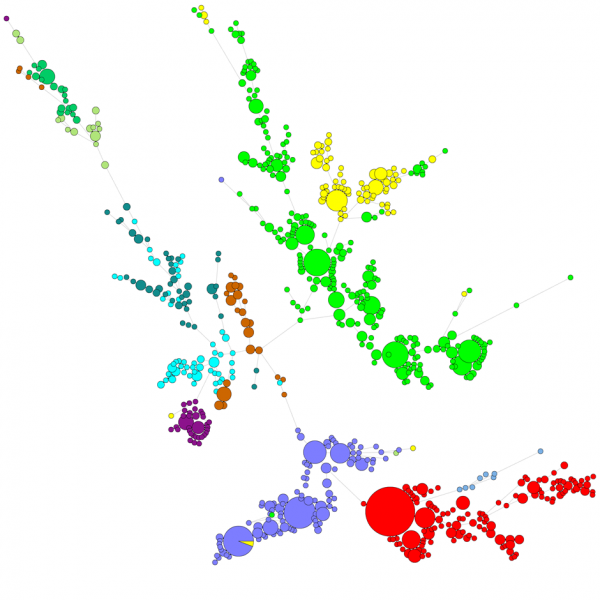
Cuando se compara un gran número de secuencias, un dendrograma UPGMA o de unión de vecinos no da una visión muy buena de los datos. En estos casos, un árbol no arraigado proporciona una mejor solución, en BIONUMERICS es posible calcular árboles de extensión mínima, árboles de máxima verosimilitud y árboles de máxima parsimonia a partir de los datos de la secuencia. Se pueden definir diferentes grupos para los datos y estos grupos se visualizan en los árboles, dando una buena visión general de las relaciones filogenéticas, el cluster presente en la base de datos y también los valores atípicos. La figura siguiente muestra un árbol de extensión mínima basado en las secuencias ITS, los grupos representan diferentes géneros.
La secuencia ITS también puede utilizarse para identificar organismos desconocidos en una base de datos. Con el módulo de clasificación e identificación, el usuario puede crear proyectos de identificación que contengan cepas identificadas y utilizarlas para identificar una cepa desconocida, se asignará una puntuación basada en la similitud junto con la fiabilidad de esta puntuación. El usuario tiene pleno control sobre los parámetros de comparación y los valores de corte para la identificación.