
Espaçadores internos transcritos (ITS) são regiões dentro da transcrição ribossômica que são excisadas e degradadas durante a maturação. As suas sequências geralmente apresentam mais variações do que a sequência ribossómica, tornando-os populares para análise filogenética e/ou identificação de espécies e estirpes. Especialmente para fungos, esta é uma técnica popular para identificação, uma vez que a identificação baseada em características morfológicas é trabalhosa e muitas vezes não leva a um resultado correcto. A região utilizada é geralmente uma combinação de ITS e sequências ribossómicas e na maioria das vezes consiste numa sequência (parcial) de 18S rRNA (16S rRNA para procariotas), uma região transcrita internamente (ITS1), a sequência completa de 5,8s rRNA, uma região transcrita internamente (ITS2) e uma sequência (parcial) de 28s rRNA. Muitas sequências ITS estão disponíveis em bases de dados públicas tais como NCBI e EBI.
ITS digitando workflow no software BIONUMERICS
Em BIONUMERICS, as sequências ITS podem ser importadas em lote a partir de ficheiros tracejados utilizando o plugin de montagem de sequências em lote. Seqüências de referência podem ser importadas de bancos de dados públicos e/ou de arquivos fasta ou GenBank.
Several algorítmos podem ser usados para realizar um alinhamento múltiplo das seqüências, Needleman-Wunsch, Wilbur-Lipman e nosso próprio algoritmo proprietário elaborado em Applied Maths. A adaptação manual do alinhamento é possível. Com base no alinhamento múltiplo, um agrupamento pode ser calculado para refletir as relações filogenéticas entre as sequências analisadas.
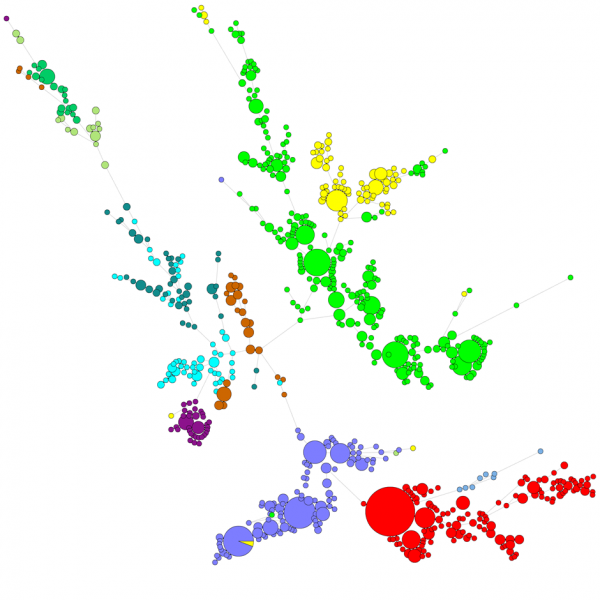
Ao comparar um grande número de sequências, um dendrograma de UPGMA ou de união de vizinhos não dá uma visão muito boa dos dados. Nestes casos, uma árvore não enraizada fornece uma solução melhor, em BIONUMÉRICA é possível calcular árvores de amplitude mínima, árvores de máxima probabilidade e árvores de máxima parcimónia a partir dos dados da sequência. Diferentes grupos podem ser definidos para os dados e estes grupos são visualizados nas árvores, dando uma boa visão geral das relações filogenéticas, do cluster presente na base de dados e também dos outliers. A figura abaixo mostra uma árvore de amplitude mínima baseada em seqüências ITS, os grupos representam diferentes gêneros.
A seqüência ITS também pode ser usada para identificar organismos desconhecidos contra um banco de dados. Com o módulo Classifiers and Identification, o usuário pode criar projetos de identificação contendo cepas identificadas e usá-los para identificar uma cepa desconhecida, uma pontuação será atribuída com base na similaridade juntamente com a confiabilidade desta pontuação. O usuário tem controle total sobre os parâmetros de comparação e os valores de corte para identificação.