
Sisäiset transkriptoidut välikappaleet (ITS) ovat ribosomaalisen transkriptin sisällä olevia alueita, jotka poistuvat ja hajoavat kypsymisen aikana. Niiden sekvensseissä on yleensä enemmän vaihtelua kuin ribosomaalisessa sekvenssissä, minkä vuoksi ne ovat suosittuja fylogeneettisessä analyysissä ja/tai lajien ja kantojen tunnistamisessa. Erityisesti sienien kohdalla tämä on suosittu tunnistustekniikka, sillä morfologisiin ominaisuuksiin perustuva tunnistaminen on työlästä eikä useinkaan johda oikeaan tulokseen. Käytetty alue on yleensä yhdistelmä ITS- ja ribosomisekvenssejä, ja se koostuu useimmiten (osittaisesta) 18S rRNA-sekvenssistä (16S rRNA prokaryoottien osalta), sisäisesti transkriptoituneesta alueesta (ITS1), 5.8s rRNA:n koko sekvenssistä, sisäisesti transkriptoituneesta alueesta (ITS2) ja 28s rRNA:n (osittaisesta) sekvenssistä. Monet ITS-sekvenssit ovat saatavilla julkisista tietokannoista, kuten NCBI:stä ja EBI:stä.
ITS:n tyypitystyönkulku BIONUMERICS-ohjelmistossa
BIONUMERICS-ohjelmistossa ITS-sekvenssit voidaan tuoda eräajona jäljitystiedostoista käyttäen batch sequence assembly -lisäosaa. Referenssisekvenssit voidaan tuoda julkisista tietokannoista ja/tai fasta- tai GenBank-tiedostoista.
Sekvenssien moninkertaiseen kohdistamiseen voidaan käyttää useita algoritmeja: Needleman-Wunsch, Wilbur-Lipman ja oma, Applied Mathsissa kehitetty algoritmimme. Kohdistus on mahdollista mukauttaa manuaalisesti. Moninkertaisen kohdistuksen perusteella voidaan laskea klusterointi, joka kuvastaa analysoitujen sekvenssien välisiä fylogeneettisiä suhteita.
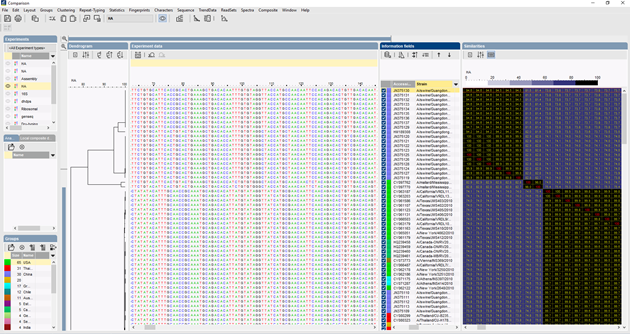
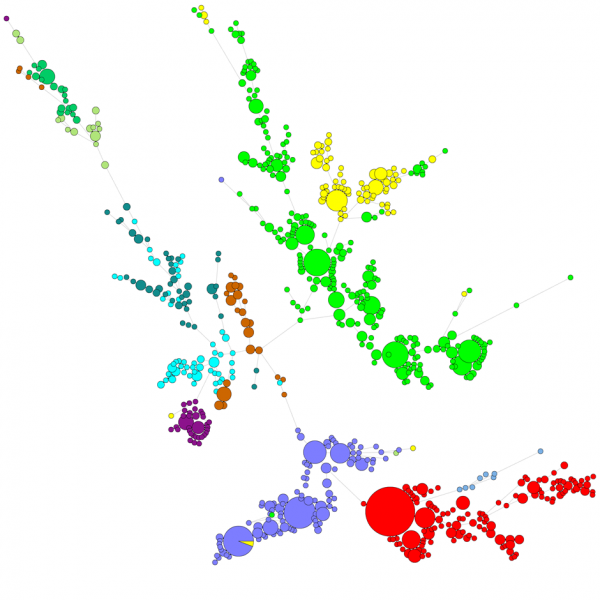
Vertailtaessa suurta määrää sekvenssejä UPGMA- tai naapuriliitosdendrogrammi ei anna kovin hyvää yleiskuvaa aineistosta. Näissä tapauksissa juurruttamaton puu tarjoaa paremman ratkaisun, BIONUMERICS-ohjelmassa on mahdollista laskea minimaalisia jännityspuita (minimal spanning trees), maksimaalisen todennäköisyyden puita (maximum likelihood trees) ja maksimaalisen parsimonian puita (maximum parsimony trees) sekvenssidatasta lähtien. Aineistolle voidaan määritellä erilaisia ryhmiä, ja nämä ryhmät visualisoidaan puissa, jolloin saadaan hyvä yleiskuva fylogeneettisistä suhteista, tietokannassa esiintyvistä klustereista ja myös poikkeavuuksista. Alla olevassa kuvassa on ITS-sekvensseihin perustuva minimaalinen kattava puu, ryhmät edustavat eri sukuja.
ITS-sekvenssiä voidaan käyttää myös tuntemattomien organismien tunnistamiseen tietokantaa vastaan. Luokittelijat ja tunnistaminen -moduulin avulla käyttäjä voi luoda tunnistushankkeita, jotka sisältävät tunnistettuja kantoja, ja käyttää niitä tuntemattoman kannan tunnistamiseen; samankaltaisuuden perusteella annetaan pistemäärä yhdessä tämän pistemäärän luotettavuuden kanssa. Käyttäjällä on täysi kontrolli vertailuparametreihin ja tunnistuksen raja-arvoihin.