
Internal Transcribed Spacers (ITS) zijn regio’s binnen het ribosomale transcript die tijdens de rijping worden uitgesneden en afgebroken. Hun sequenties vertonen over het algemeen meer variatie dan de ribosomale sequentie, waardoor zij populair zijn voor fylogenetische analyse en/of identificatie van soorten en stammen. Vooral voor schimmels is dit een populaire techniek voor identificatie omdat identificatie op basis van morfologische kenmerken omslachtig is en vaak niet tot een correct resultaat leidt. De gebruikte regio is meestal een combinatie van ITS- en ribosomale sequenties en bestaat meestal uit een (gedeeltelijke) 18S rRNA-sequentie (16S rRNA voor prokaryoten), een intern getranscribeerd gebied (ITS1), de volledige sequentie van 5,8s rRNA, een intern getranscribeerd gebied (ITS2) en een (gedeeltelijke) sequentie van 28s rRNA. Veel ITS-sequenties zijn beschikbaar in openbare databases zoals NCBI en EBI.
ITS-typeringsworkflow in de BIONUMERICS-software
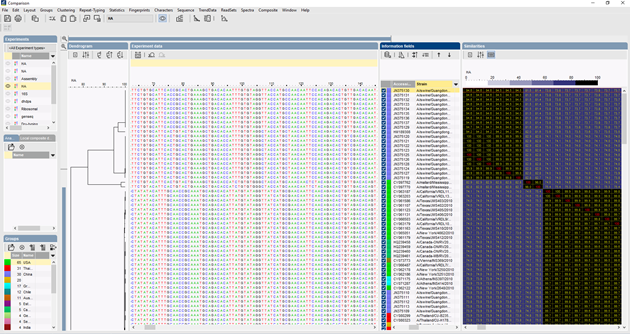
In BIONUMERICS kunnen de ITS-sequenties in batch worden geïmporteerd vanuit trace-bestanden met behulp van de batch sequentieassemblage-plugin. Referentiesequenties kunnen worden geïmporteerd uit openbare databases en/of uit fasta- of GenBank-bestanden.
Er kunnen verschillende algoritmen worden gebruikt om een meervoudige alignment van de sequenties uit te voeren, Needleman-Wunsch, Wilbur-Lipman en ons eigen algoritme dat bij Applied Maths is uitgewerkt. Handmatige aanpassing van de uitlijning is mogelijk. Op basis van de meervoudige uitlijning kan een clustering worden berekend die de fylogenetische relaties tussen de geanalyseerde sequenties weergeeft.
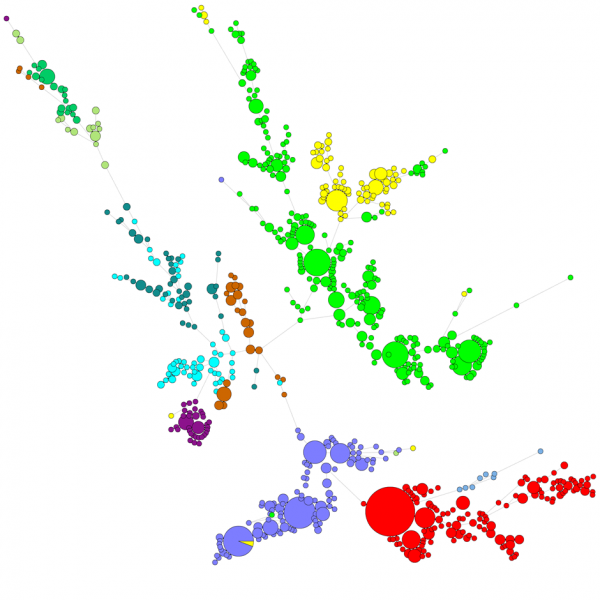
Wanneer een groot aantal sequenties wordt vergeleken, geeft een UPGMA- of neighbor-joining dendrogram geen erg goed overzicht van de gegevens. In deze gevallen biedt een boom zonder wortels een betere oplossing. In BIONUMERICS is het mogelijk om op basis van sequentiegegevens minimal spanning trees, maximum likelihood trees en maximum parsimony trees te berekenen. Verschillende groepen kunnen gedefinieerd worden voor de data en deze groepen worden gevisualiseerd in de bomen, wat een mooi overzicht geeft van de fylogenetische relaties, de cluster aanwezig in de database en ook de uitbijters. Onderstaande figuur toont een minimaal overspannen boom op basis van ITS-sequenties, waarbij de groepen verschillende genera vertegenwoordigen.
De ITS-sequentie kan ook worden gebruikt om onbekende organismen te identificeren aan de hand van een database. Met de Classifiers- en Identificatiemodule kan de gebruiker identificatieprojecten aanmaken met geïdentificeerde stammen en deze gebruiken om een onbekende stam te identificeren; er wordt een score toegekend op basis van de gelijkenis samen met de betrouwbaarheid van deze score. De gebruiker heeft volledige controle over de parameters voor vergelijking en de cutoff-waarden voor identificatie.