
Gli spaziatori trascritti interni (ITS) sono regioni all’interno del trascritto ribosomiale che vengono eliminate e degradate durante la maturazione. Le loro sequenze mostrano generalmente più variazioni rispetto alla sequenza ribosomiale, rendendole popolari per l’analisi filogenetica e/o l’identificazione di specie e ceppi. Specialmente per i funghi, questa è una tecnica popolare per l’identificazione poiché l’identificazione basata sulle caratteristiche morfologiche è laboriosa e spesso non porta a un risultato corretto. La regione usata è di solito una combinazione di sequenze ITS e ribosomiali e il più delle volte consiste in una sequenza (parziale) di 18S rRNA (16S rRNA per i procarioti), una regione internamente trascritta (ITS1), l’intera sequenza di 5.8s rRNA, una regione internamente trascritta (ITS2) e una sequenza (parziale) di 28s rRNA. Molte sequenze ITS sono disponibili su database pubblici come NCBI e EBI.
Flusso di lavoro di tipizzazione ITS nel software BIONUMERICS
In BIONUMERICS, le sequenze ITS possono essere importate in batch da file di traccia utilizzando il plugin di assemblaggio sequenze batch. Le sequenze di riferimento possono essere importate da database pubblici e/o da file fasta o GenBank.
Possono essere utilizzati diversi algoritmi per eseguire un allineamento multiplo delle sequenze, Needleman-Wunsch, Wilbur-Lipman e il nostro algoritmo proprietario elaborato da Applied Maths. L’adattamento manuale dell’allineamento è possibile. Sulla base dell’allineamento multiplo, un clustering può essere calcolato per riflettere le relazioni filogenetiche tra le sequenze analizzate.
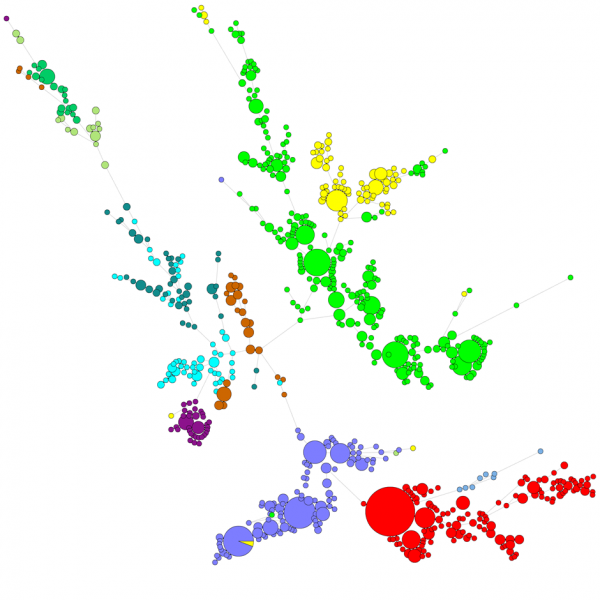
Quando si confronta un gran numero di sequenze, un UPGMA o un dendrogramma neighbor-joining non dà una visione molto buona dei dati. In questi casi, un albero non radicato fornisce una soluzione migliore, in BIONUMERICS è possibile calcolare alberi di minima estensione, alberi di massima verosimiglianza e alberi di massima parsimonia partendo dai dati di sequenza. Diversi gruppi possono essere definiti per i dati e questi gruppi sono visualizzati negli alberi, dando una bella panoramica delle relazioni filogenetiche, i cluster presenti nel database e anche gli outlier. La figura qui sotto mostra un albero minimal spanning basato sulle sequenze ITS, i gruppi rappresentano diversi generi.
La sequenza ITS può anche essere usata per identificare organismi sconosciuti rispetto a un database. Con il modulo Classificatori e Identificazione, l’utente può creare progetti di identificazione contenenti ceppi identificati e usarli per identificare un ceppo sconosciuto, un punteggio sarà assegnato in base alla somiglianza insieme all’affidabilità di questo punteggio. L’utente ha il pieno controllo sui parametri di confronto e sui valori di cutoff per l’identificazione.